2023年,对于很多制药企业来说,是充满挑战和机遇的一年。很多制药企业不断地传来捷报,当然也有很多品种不被批准。对于捷报,大家都会分析借鉴,但不批准的品种同时也是一朵朵美丽的花,成功也许很难复制,但对失败的认真分析和复盘总会带来收获!
在中国仿制药企业工作,老板最关注的就是什么时候有成果,什么时候能获批?获批后能不能进集采,利润有多少?笔者研究了集采、上市、中标的要求,发现一致性评价和视同通过一致性评价的品种,是获得上市销售的基本要求,对比了一致性评价和仿制药的受理后的审评时间,现分享给大家:
一、一致性评价申请是弯道超车的一条捷径
首先CDE对审评时限的要求为:
笔者根据上述审评时间进行整理,见表1:
▲表1-仿制药和一致性评价受理至获批时限考察分析
丁香园发布的《2023年度化药和生物制品审评报告》
从上表可知,实际申报时间统计与审评时限还是很接近的,受理到获批实际时间稍微长一些,最主要的是两个原因:
1.发补以后提交二次检验;
2.发补审核完成后需要进行生产工艺信息表、说明书和质量标准复核等。
此外,山东华鲁制药有限公司按照一致性评价的新增规格的4ml:500mg(受理号为CYHB2350093)于2023年1月30日受理,于2023年10月30日获批,仅花费了9个月。
综上,不管是从法规时限还是从实际品种受理到获批的时限来看,仿制药都比一致性需要多6个月,对于有批文可以进行一致性评价的企业来说是比较友好的。
二、一致性评价的不批准原因分析
虽然一致性评价有其法规方面审评时限的优势,但是因为企业自身原因而导致优势不能充分发挥也是屡见不鲜。为此,笔者对一致性评价不批准的原因进行了充分的调研分析,以便读者借鉴。
2024年2月8日,CDE网站上发布的“2023年药审报告划重点:IND、NDA申请双旺盛,以患者为中心理念首次纳入指导原则”一文中指出,2023年,药审中心首次将“以患者为中心”和基于“动物法则(AnimalRule)”药物注册理念纳入指导原则,标志着我国药物研发策略进入了新阶段。据悉,“以患者为中心”的药物研发是指基于患者角度开展的药物开发、设计、实施和决策的过程,旨在高效研发更符合患者需求的有临床价值的药物,是当前各国药品监管机构积极探索的领域。
在不批准的领域里,立题合理性和临床合理用药等方面是CDE重点关注的对象,本章节整理了国家药品通知件中的一致性评价品种,并对整理的品种进行调研,选择可以看出不批准原因的品种进行浅显的分析,不当之处,希望各位同仁给予指导。
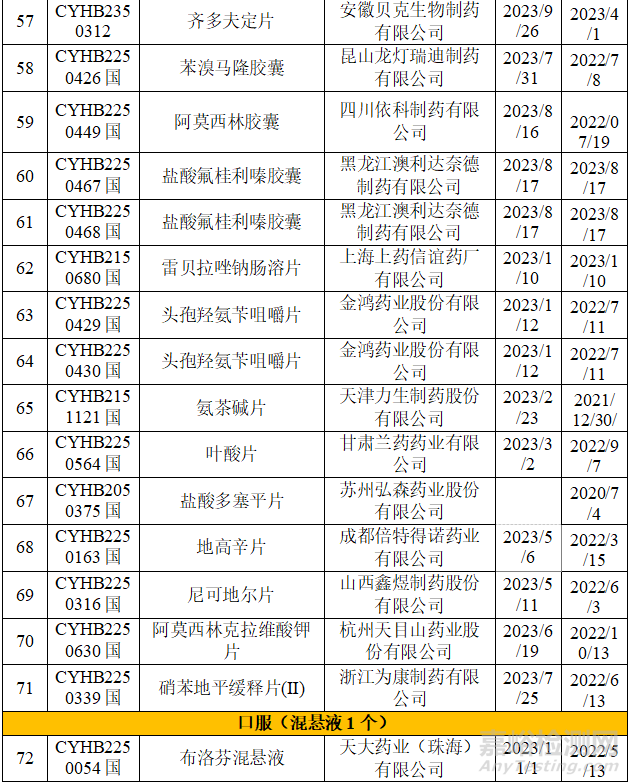
从上表可知,2023年药品通知件中共72个受理号,注射剂53个,占73.6%,其中注射液31个,冻干粉针22个;口服固体制剂18个,混悬剂1个。归结起来主要有两个原因:1.参比制剂;2、规格的临床合理性。
2.1 参比制剂一直未公布,审评结论不通过
盐酸托烷司琼注射液,西南药业股份有限公司于2019年申报,参比制剂一直未公布,在参比制剂第63批中,给予参比制剂不通过的结论:“经一致性评价专家委员会审议,为确保参比制剂的质量,建议参比制剂首选欧盟、美国以及日本等监管体系较为完善的机构批准上市的原研药品,审议未通过”。给予笔者的反思:对于参比制剂,优选欧美日上市的品种,笔者对这个品种进行初步调研,发现确实存在欧洲上市的品种。
2.2 参比制剂调出
2023年9月18日,CDE官网再次发布将该参比制剂调出参比制剂目录的征求意见稿,内容与第四十六批_征求意见稿调出品种目录内容一致,具体调出原因为:经一致性评价专家委员会审议,拟调出参比制剂因采用终端灭菌工艺制备的奥沙利铂注射液可以满足产品杂质控制及稳定性要求,奥沙利铂注射剂应首选终端灭菌的注射液,注射用奥沙利铂为不合理剂型,予以调出。因此将一直没有给出结论的扬子江药业和恒瑞申报的奥沙利铂注射剂一致性评价给予不批准的结论。
按照CDE 2020年发布的《化学药品注射剂灭菌和无菌工艺研究及验证指导原则(试行)》,注射剂只要有终端灭菌工艺,则其他非终端一定不会被批准。这是属于不批准的红线,提醒各位谨慎立项非终端灭菌工艺的品种。
2.3 临床规格用量合理性
2.3.1 注射用阿奇霉素
参比制剂的规格为0.5g,而国内原有规格中有0.125g(25个文号),0.25g(50个文号),在进行一致性评价时,由于两种规格小于临床单次使用的最小剂量,而不被批准。
2.3.2 注射用更昔洛韦
公布的参比制剂规格为0.5g,武汉人福申报的0.25g和0.5g两个规格均通过一致性评价,而申报的0.05g的规格一致性评价未通过。因此,大家在立项时一定要考虑临床的单次最小使用量。
2.3.3 胞磷胆碱钠注射液
公布的参比制剂规格为4ml:500mg和4ml:1g;山东齐鲁制药有限公司申请了原有品种规格2ml:0.25g的一致性评价未获批。
三、新药品种不批准的品种统计
近年来,多重医药创新鼓励政策持续激发,产业创新活力持续释放,2023年药品注册申请申报量持续增长。我们在看到创新药给企业带来利好的同时,也应看看那些不批准的品种的情况,笔者根据国家局药品通知件待领取信息进行整理,共整理了30个不批准的创新药,其中化1类:10个;2类9个;5.1类11个。具体信息见表3:
▲表3-2023年创新药品种不批准的品种汇总
对新药的查询让笔者对坚持做创新药的企业肃然起敬,每个品种都经历了漫长的立项、临床试验和最终的获批或者不获批的情况。
首先09年申报的硫酸普拉睾酮钠片。该品种经历过药物临床试验数据自查核查品种(2015.7.22);于2016年10月13日开展临床试验,2019年终止试验。经历了10多年的时间,是一代制药人的坚持。
对于新药不获批的情况,笔者归结的原因有不能满足临床需要、技术壁垒、原料撤回等,对新药的不批准的分析如下:
3.1 临床试验是否完善
2.4类为增加国内外均未有的适应症,临床试验的结果是影响其能否获批的主要原因。
3.2 技术壁垒
齐鲁制药的他达拉非口溶膜与他达拉非片进行BE试验,获批生产。而恒瑞的同品种在进行BE试验后直接申请上市,未取得发补机会,直接给予不批准的命运。笔者反思认为:对于药品来说,技术壁垒和药学质量一直是产品的核心,许多人认为临床等效了就肯定能获批而忽略了药品本身技术的壁垒。
3.3 得原料者得天下
3.3.1 羧胺三唑软胶囊
该原料在原料备案平台上,仅珠海润都制药股份有限公司进行登记,目前状态为I;
3.3.2 注射用普那布林浓溶液
该品种的原料药登记厂家为凯莱英医药集团(天津)股份有限公司,目前原料药登记状态仍然为I。因此,大家在做新药原料加制剂的时候,原料药的质量一定要把控,可以找一些第三方咨询公司进行一下评估。所有的新药都是要经历临床研究的,花费都是比较多的,最后因为原料而不批准,就得不偿失了。
3.3.3 利拉鲁肽注射液——原料竞争
利拉鲁肽注射液申报按照化学药进行申报,其他获批的是按照生物药进行申报的。查询CDE原料药备案平台,利拉鲁肽的原料的备案信息均为I。该品种按照化学药申报,转化为原料获批的竞争了。就像司美格鲁肽一样,原研是发酵,仿制是化学合成,但美国也有处于A状态的DMF了。不管是化药合成还是生物发酵,谁能够将原料做出来并得到官方认可,谁就能拿到这个品种的市场。不管是按照化药还是生物药仿制,必要的安全性研究和相应的临床试验一样不能少,只有这样才能拿到批件而非药品通知件!