您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2024-09-24 09:01
根据日本《药品与医疗器械法》(Pharmaceutical and Medical Device Act,缩写为PMD Act),日本的药品和医疗器械管理是由卫生劳动和福利部(日本厚生省)(Ministryof Health, Labor and Welfare,MHLW)负责。
由于主管机构的英文缩写是PMDA,业内把日本医疗器械合规过程通称为PMDA认证(也有人称为MHLW认证)。
一、PMDA和MHLW
日本的药品和医疗器械管理是由卫生劳动和福利部(日本厚生省)(Ministryof Health, Labor and Welfare,MHLW)负责。
PMDA全称为Pharmaceuticals and Medical Devices Agency(独立行政法人药品和医疗器械综合机构),是MHLW管辖的独立行政法人。PMDA的业务包括审查、安全对策、健康损害救济等,对医疗器械进行技术复核和相关研究工作。
在日本,药品、医疗器械管理法律法规主要分为三类:
● 由日本议会批准通过的称法律;
● 由日本政府内阁批准通过的称政令或法令;
● 由厚生省大臣批准通过的称告示或省令。
二、医疗器械的等级分类
日本医疗器械术语集(Japanese Medical Device Nomenclature , JMDN) 编码明确了器械分类与注册登记路径。根据风险等级对医疗器械实施分类管理,按照风险等级从低到高分为4个等级。
1.Class I:风险极低的医疗器械(一般医疗器械,如手术刀)
由日本厚生劳动省大臣在听取“药事和食品卫生审议会”的意见后进行指定,这类医疗器械在出现副作用或功能损害时,对人的生命和健康影响的风险很小。一般指除高度管理医疗器械和管理医疗器械以外的医疗器械。 须进行地方政府备案,无实质性审查。
2.Class II:风险较低的医疗器械(受控医疗器械,如电子内窥镜、消化器官用导管)
由日本厚生劳动省大臣在听取“药事和食品卫生审议会”的意见后进行指定,指除高度管理医疗器械以外的医疗器械。这类医疗器械在出现副作用或功能损害时,有可能影响人的生命和健康,需要进行合适的管理。Ⅱ类医疗器械实施上市前认证(Ninsho)。特殊受控 II 类医疗器械上市前需要由医疗器械上市许可持有人提交上市前认证,认证机构需要得到PMDA授权。须由第三方认证机构RCB负责审查。
3.Class III、Class IV:风险较高或者极高的医疗器械(高度受控医疗器械,如透析器、人工骨骼、人工呼吸器、心脏血管用球囊导管,起搏器、人工心脏、支架等)
除特殊受控 II 类医疗器械外的其他Ⅱ类、Ⅲ类和Ⅳ类医疗器械必须由医疗器械上市许可持有人向PMDA 提交上市前批准申请,经PMDA 批准后才能投放市场。须进行PMDA审查。
三、备案或认证模式
1.classⅠ类医疗器械上市前必须由医疗器械上市许可持有人。向PMDA 提交一份上市前文件即产品备案,该文件不需要经过PMDA 的审核和批准。
2.class Ⅱ类医疗器械实施上市前认证(Ninsho)。特殊受控 II 类医疗器械上市前需要由医疗器械上市许可持有人提交上市前认证,认证机构需要得到PMDA授权(即有资质的第三方认证机构。)
3.classⅢ类、classⅣ类医疗器械实施上市前批准(Shonin)。除特殊受控 II 类医疗器械外的其他Ⅱ类、Ⅲ类和Ⅳ类医疗器械必须由医疗器械上市许可持有人向PMDA 提交上市前批准申请,经PMDA 批准后才能投放市场。
四、具体注册流程
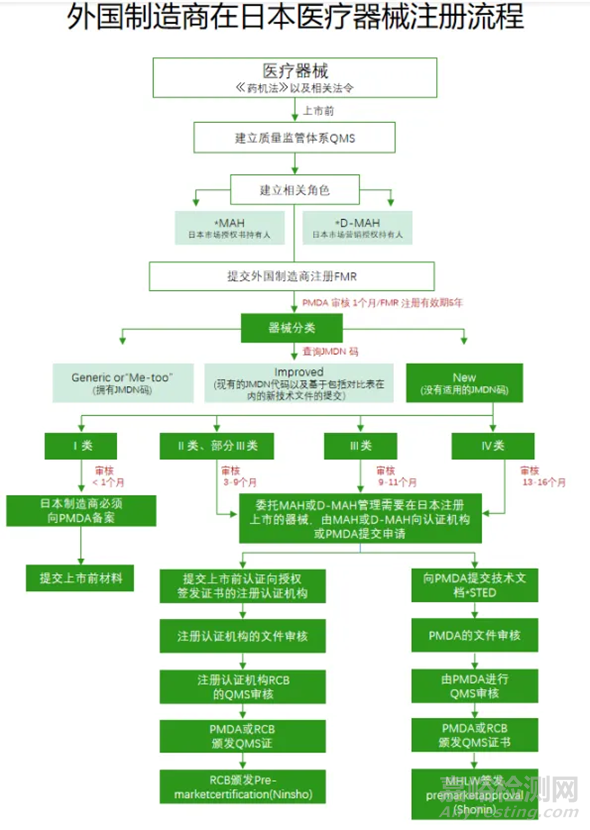
重点关注:
所有类别器械:任命MAH或D-MAH管理日本器械上市前申请或审批。
MAH全称Marketing Authorized Holder(日本上市许可持有人),拿到某一类产品MAH执照后,才可以提出具体产品的上市申请。由于外国公司在日本没有办事处,需要任命一名在日本持有营业执照的指定上市许可持有人D-MAH (Designated Marketing Authorization),协调货物放行给外国公司的经销商以及处理投诉和警戒信息事宜。
外国制造商必须指定日本国内市场授权持有人,这是作为在日本国内销售医疗器械的首要条件。MAH对日本市场上的医疗器械产品从批准、认证和备案,到上市后的安全管理负有最终责任,并对问题产品实施召回,且有义务管理和监督产品生产。
除MAH外,PMDA允许外国制造商指定市场营销授权持有人,即D-MAH。D-MAH不拥有产品的注册和证书/批准的控制权,可视作为外国制造商驻日代表。
五、STED摘要(注册资料)
01产品规格
02稳定性和保质期数据
03性能测试数据
04风险分析
05临床数据
06制造(过程,监督,灭菌)
07遵守适用的标准和基本原则
08开发历史记录(先前的设备版本,全局授权)
六、质量管理体系(QMS)
QMS审核是由医药品与医疗器械局(PMDA)或某一家注册过的认证机构(RCB)来执行的。QMS审核所需资料有质量手册、程序文件、ISO 13485证书、组织结构图、制造工程图、制造设备等。
QMS审核范围包括制造销售商(MAH)、医疗器械的设计、生产制造相关的所有场所。

来源:Internet


