您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-12-02 09:06
分析方法生命周期是一个持续的开发过程,其建立在对候选新药的发现、临床前和临床开发过程中使用的每种分析方法的了解之上。分析方法生命周期是分析方法定义、开发、优化、验证、鉴定、确认、转移、维护以及最终报废的流程图。生命周期的综合方法必须确保分析方法从最初的设计和开发发展到商业使用,并且必须在用于商业化道路上的各个临床阶段之前证明适合其预期目的。对分析方法进行评估和选择,以确定药物的具体特征。开发和验证分析测定方法,以根据预先确定的验收标准测试药物和药物产品的具体特征。
分析方法生命周期方法强调,在开发、控制、建立和使用分析程序时,必须有健全的科学流程和质量风险管理。这种方法适用于所有类型的分析方法,所付出的执行程度应与技术的复杂性和所要测量的质量属性的关键性相一致。熟练程度和技术知识是分析方法开发的基本要求,而要构建分析生命周期工作流程以满足预期药物使用的监管要求,则需要深厚的质量和监管知识。图 14-1 表示分析方法生命周期的关键要素及其与产品开发的联系。
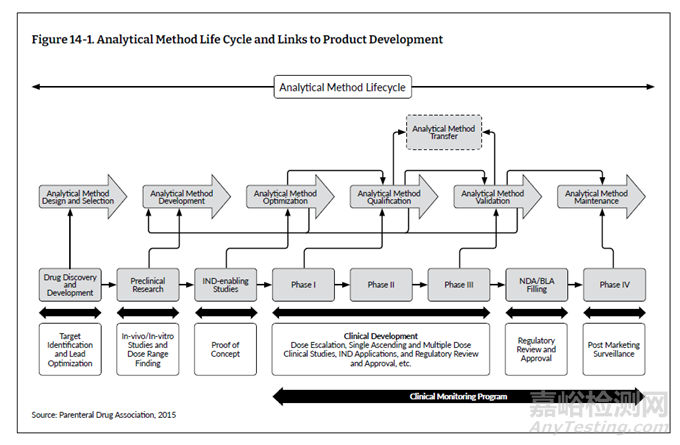
1、分析方法定义:选择与优化
方法开发流程的第一步是根据预期用途定义方法的适用范围、目的和目标,以确保方法设计、功能和性能预期保持一致。在此阶段,需要制定协议来定义分析开发计划。分析方法开发的预期用途包括表征、过程测试、常规发布测试、稳定性、生物利用度、原料发布或过程验证支持。
预期用途与产品开发阶段直接相关,从临床前到临床再到商业阶段。在方法定义阶段确定测试样品也很重要。测试样品的例子包括原料药(DS)、制剂(DP)、安慰剂、基质(例如配方缓冲液、生物液体)、活性药物成分、赋形剂、杂质等。检测类别根据国际人用药品技术要求协调委员会(ICH)Q2(R1)指南中关于鉴别、定量杂质、极限测试或检测含量/效力的规定进行定义。分析开发计划的目标可以是正交、表征、可比性、放行、鉴定或鉴定和验证、稳定性或生物利用度研究。建议采用更全面的方法来定义分析方法,以降低方法开发计划与最终方法是否适合其预期用途之间不匹配的风险。
1、分析目标简介:分析方法生命周期结构框架
质量先驱约瑟夫·M·朱兰(Joseph M. Juran)博士提出了“质量源于设计”(Quality by Design (QbD))的概念。珍妮特·伍德科克博士将高质量药品定义为无污染、能够可靠地向消费者提供标签上承诺的治疗效果的产品。ICH Q8指南强调了QbD概念,即设计最终产品的质量,而不是测试产品。ICH Q8将QbD定义为“一种系统化的开发方法,从预先确定的目标开始,强调基于可靠科学和质量风险管理的对产品和工艺的理解以及过程控制”。FDA鼓励在药品开发、生产和监管中采用基于风险的方法和QbD原则。FDA之所以重视QbD,是因为认识到增加测试并不一定能提高产品质量。质量必须融入产品中(Quality must be built into the product)。QbD要求对药品的预期质量特性进行前瞻性总结。
质量目标产品概况(Quality Target Product Profile ,QTPP)是对药品质量特性的前瞻性总结,在考虑药品的安全性和有效性后,理想情况下应达到这些质量特性,以确保达到预期的质量。QTPP构成了产品设计和开发的基础。QTPP推动了产品质量特性的定义。分析目标概况(Analytical Target Profile ,ATP)是分析方法的同义词。
在确定药品的分析目标概况(ATP)时,必须记录ICH Q2(R1、R2)类别分析方法(analytical methods)、特性(identity)、纯度(purity)、杂质(impurity)和含量(assay)以及性能特征(如准确性、精确性和特异性)的预定义方法参数。ATP中定义的分析测试程序和可报告结果决定了产品的质量属性,并依次为整体控制策略提供关键输入。分析目标概况(ATP)指导着适当分析方法的设计和开发。
分析目标概况(Analytical Target Profile ,ATP)是提供性能标准的基础,用于交付优质产品,并支持分析方法的“符合目的”或“适合预期用途”声明。用于测量关键质量属性(CQA)的分析方法必须足够灵敏、准确和精确,以便对属性进行充分控制,无论该方法用于过程控制还是通过DS或DP的最终产品规格。
定义阶段需要确定ICH Q2(R1)中规定的用于预期用途的纯度、杂质和检测的分析方法,确定关键质量属性(CQAs),确定方法性能的质量属性,根据支持信息确定每个属性的风险分配,以及方法性能的具体可接受性标准,例如系统适用性、检测可接受性、基于生产量的样品可接受性、设备、可转移性、材料和试剂可用性以及检测关键性。
分析方法定义阶段结束后,可以生成一份分析目标概况(Analytical Target Profile ,ATP)报告。根据产品在开发生命周期中的阶段,ATP报告可以是一个独立的交付物,也可以是最终方法开发报告的关键要素。随着新技术的出现、根据法规反馈对方法提出新要求或流程变更,ATP报告可能会被更新并用于指导分析生命周期管理中的进一步方法开发工作。
2、分析方法选择
一旦确定了分析目标概况(Analytical Target Profile ,ATP),分析方法生命周期的下一步就是选择适合目的或预期用途的方法。方法选择应包括对各种技术的评估,这些技术可应用于测试要求并满足产品规格的法规要求。方法的选择基于分析化学,其中包括识别、分离和量化药物化合物化学成分的方法。选择阶段包括选择材料和试剂、样品、设备和实验室。在确定是内部分析还是将分析方法外包给第三方实验室测试以满足法规要求时,用户需求、实验室能力和预算分配起着重要作用。选择阶段需要交付的是符合产品预期用途法规要求的方法选择建议,以及方法开发或方法优化策略的最终确定。
出于记录和存档的目的,可以生成与ATP类似的方法选择和策略报告。该报告可包含以下部分:
• 项目名称
• 项目发起人/主要研究者(如适用)
• 产品开发阶段(临床前、临床研究性新药(IND)阶段、1期、2期或3期
• 应用(放行、稳定性、in-process、生物利用度)
• 产品基质(例如配方缓冲液)
• 分析物基质(例如生物液体、配方缓冲液)
• 预期用途的法规要求[例如,良好实验室规范GLP、良好临床规范GCP、现行良好生产规范(cGMP)]
ICH Q2(R1)规定的分析方法类型
• 拟议规范
• 样品可接受性标准
• 系统适用性标准
• 拟采用的方法及其选择依据
• 替代方法
• 可能出现的短期和长期问题(例如供应链短缺)
3、分析方法的开发与优化
分析方法开发(Analytical method development AMD)是任何药物开发项目不可或缺的组成部分。方法开发是分析方法生命周期中的重要里程碑。分析方法开发的目的是建立分析目标概况(ATP)中规定的药物安全性、特性、强度、纯度和效力的最佳条件。分析方法开发的范围包括选择和优化实验设计以及评估方法性能。方法开发和优化的目标是选择合适的方法参数。交付成果可以是生成开发报告和标准操作程序草案(SOP)。
方法开发提供了关键信息,用于评估该方法是否适合目的,并可根据其预期用途进行方法鉴定或验证。分析开发报告是一份包含方法性能详情、材料和试剂信息、参考标准、对照、系统适用性要求、方法获取和数据分析参数、设备和软件的文档。对于任何开发新药候选物的制药或生物技术公司而言,分析方法开发都是工艺开发的关键支柱。
可采用分阶段的方法来满足法规对方法开发水平的期望。该方法在可靠性和便利性方面进行了优化,可根据其在产品开发阶段的应用情况采用增量开发和优化方法。例如,在产品开发的早期阶段(即临床前和1期),高效液相色谱法可能专注于持续测量目标分析物的含量、纯度和稳定性,但可能无法提供色谱图中所有杂质的特性和响应因子。然而,在产品研发后期(例如2期和3期),同样的方法可以提供这些信息,并将杂质标准纳入分析方法。
方法开发包括优化提取和检测目标分析物的程序和条件。它包括优化以下参数,以确保该方法适用于鉴定或验证(qualification or validation):
• 参考标准(Reference standards)
• 关键试剂(Critical reagents)
• 校准曲线(Calibration curve)
• 质量控制(QC)样品(Quality control (QC) samples)
• 选择性和特异性(Selectivity and specificity)
• 灵敏度(Sensitivity)
• 精确度(Accuracy)
• 精确度(Precision)
• 回收率(Recovery)
• 基质中分析物的稳定性(Stability of the analyte in the matrix)
在分析方法生命周期中,结束方法开发阶段所需的关键成果是方法开发报告和标准操作程序草案。根据公司政策和程序,分析方法开发(Analytical method development AMD)报告的格式可能有所不同,但必须提供所有开发研究、数据、实验和结论的信息,以支持分析方法生命周期的下一步。该报告是建立标准操作程序和鉴定或验证协议目标标准的基石。与AMD报告一起,材料供应商、设备升级、缓冲液体积增加、关键材料属性、历史数据和趋势等信息构成了AMD包。对AMD包进行技术评估,以确定开发数据是否足以支持方法鉴定或方法验证研究。
AMD包可能包含以下可交付成果:
• 方法的目的和范围
• 设计和开发历史
• 所选方法的科学原理背景描述
• 评估过的参数和性能
• 开发步骤总结
• 目标样本和基质
• ICH Q2(R1)方法类别
• 纳入开发目标中的ICH Q2(R1)特性
• 压力研究Stress studies(如果在开发阶段进行了初始力衰减研究)
• 正在进行的长期研究的稳定性数据
• 系统适用性、方法和样本可接受性标准
• 实现预期用途
• 实现方法策略
• 材料、试剂、标准和对照
• 关键材料属性(critical material attributes,CMA)的识别
• 趋势数据和历史结果
• 实验室对产品预期用途的法规要求
• 操作员资质和培训要求
• 起草标准操作流程
4、分析方法鉴定
分析方法确认(AMQ)是业内公认且广泛接受的方法,用于证明分析方法在科学上合理、符合目的或适合预期用途、一致且能够成功生成可靠的结果。AMQ根据ICH Q2(R1)指南(即准确性、精确性、线性、特异性、范围、检测限/定量限和稳健性)确定与分析方法验证(Analytical Method Validation,AMV)相似的方法性能质量属性,但以阶段适当的方式推进,并与方法的预期用途保持一致。AMQ和AMV都证明了该方法适用于其预期用途,但它们在研究稳健性方面有所不同。
这意味着,与1期研究相比,对方法性能或资格进行更全面的评估可能有助于2期临床试验研究。与阶段相适应的监管指导文件描述了针对最终产品(DS或DP)发布的资格和验证研究的要求,以期用于预期的临床阶段。在分析方法验证(AMV)之前,分析方法确认(AMQ)并非法规要求,但它是ATP中定义的成功验证和方法适用性要求的基础。非营利组织(如肠外药物协会)发布的技术报告为AMQ提供了非约束性指导,并得到制药公司和合同分析测试实验室的广泛认可,以满足临床阶段对基于预期用途的分析方法的适当期望。
在分析方法生命周期中,AMQ可以是流程特性、可比性研究、用于在规格外调查期间确认根本原因的正交特性方法等活动的最终目标,以支持产品放行和产品开发材料的放行 /稳定性测试(即Phase 1和Phase 2临床试验材料)。根据产品所处的阶段以及开发生命周期中的分析方法,可能需要进行部分方法验证或交叉方法验证。方法学验证可作为依据ICH Q2(R1)指南进行完全方法学验证的基础。
监管机构发布了关于提交临床试验研究档案要求的指导方针。这些指导方针并未详细描述临床阶段适用的分析方法要求,但为AMQ对预期产品用途的预期奠定了基础。这些指导文件包括但不限于:
• 美国1期和2/3期临床试验的IND申报
• 在欧洲进行临床试验的研究用药品档案提交
随着产品和分析方法在开发周期中的发展,分析方法也会进行科学或技术改进。在对方法进行重大修改后,以及在将其用于临床研究之前,需要评估方法重新认证的必要性。
方法鉴定报告包括结果、评估以及关于方法是否适用于预期用途的结论。AMD数据包附在AMQ报告中。鉴定后分析方法审查的责任对于保持方法的合格状态非常重要。
这些活动包括确保方法的使用符合预期用途,并确保其符合分析生命周期开发和鉴定阶段所定义的目的。鉴定后的变更可能包括新的参考标准或对照样品评估、基于产品开发阶段的范围变更,或将方法的技术转让给其他小组或实验室。
药典方法在使用前不需要进行确认,我们将在本章后面讨论。
AMQ实验和结果的关键交付物在AMQ报告中进行了总结。在AMQ实验完成并起草了鉴定报告后,起草了SOP并将其转移至QC实验室。分析方法鉴定是分析方法和产品开发生命周期中的关键成就。AMQ报告包括:
• 设备的校准状态
• 设备软件和版本、采集方法和数据分析
• 材料、试剂、标准品、对照品和样品
• 样品制备和采集方法的描述
• 数据分析和计算方法
• 所有数据文件的参考资料,可清晰追溯到原始数据源
• 每个鉴定协议中预先定义的鉴定要素,以及根据目标标准进行的评估,并附有结论
• 技术评估,以确定每个鉴定要素的验收标准
• 意外结果和降低风险措施
• 关键方法评估(CMA),如确定
• 必要时,确保结果可重复性的关键方法步骤
• 资格鉴定过程中使用的SOP草案或版本的副本
• 根据鉴定结果,对标准操作程序(SOP)的修改建议、说明和理由
• AMD 报告和 ATP(附于 AMQ 报告之后)
2、分析方法验证
分析测试方法验证实验可提供关键数据,支持任何根据法规要求生产的药物的安全性、特性、强度、效力和纯度。测试结果对于确保消费者的健康和安全至关重要。因此,在现行cGMP下用于药品特征分析的方法应在产品生命周期中尽早验证。作为分析方法验证的一部分,所获得的数据提供了书面证据,证明测试程序适用于其预期用途,是方法性能的客观证据,并确保结果的质量和可重复性。
1、法规要求和指导文件
ICH Q2(R1)为分析方法验证(AMV)研究提供了指导并奠定了基础,以确保产品的安全性、有效性和质量。此外,ICH Q7、FDA指导文件、美国和欧盟药典以及技术报告指南对AMV进行了详细描述。
分析方法验证流程因负责药物研发和产品的预期用途的监管机构而异。法规并未对AMV进行详细说明,因此制药公司和合同分析测试实验室需要依靠监管机构和政府当局发布的指南来满足合规性要求。根据《分析程序验证:文本和方法 Q2(R1)中,AMV的关键要素包括准确性、精确度(可重复性、中间精度、可再现性)、特异性、检测限、定量限、线性、范围和稳健性。
监管指南包含不具约束力的建议,但符合监管要求,并提供有关方法验证的官方参考。可用指导文件的示例包括:
• 分析程序验证:文本和方法 Q2(R1),ICH 2005
• 行业指南:药物和生物制剂的分析程序和方法验证,FDA 2015
• 行业指南:生物分析方法验证(指南),FDA 2018
• <1225> 药典程序验证,USP 41-NF 36
• <1226> 药典程序验证,USP 41-NF 36
• <1058> 分析仪器鉴定,USP 41-NF 36
• FDA关于流程验证的指南
• EMA关于流程验证的指南
• ICH M4Q关于通用技术文件(CTD)模块3——质量部分
• ICH Q5C稳定性测试
• ICH Q6 关于测试程序和验收标准
• ICH Q8-11,关于药品开发、风险管理和质量体系
• 欧洲药品质量管理局(EDQM)关于分析程序验证/确认的指南 PA/PH/OMCL (13) 82 R5
• EMA生物分析方法验证指南
• OECD《单一实验室定量分析方法验证指导文件》
分析方法验证(AMV)实验和结果的交付物在验证报告中进行了总结。AMV报告的组成部分与AMQ报告类似,但实验的范围和深度旨在满足适当的预期用途。应在ICH M2电子通用技术文件(eCTD)新药申请(NDA)/生物制品许可申请(BLA)规范中相应部分提交记录完整的AMV报告。在产品生命周期中,随着生产工艺的发展或变化,应考虑采用基于风险的方法对现有分析方法进行重新验证。
当分析方法作为新药申请(NDA)、仿制药申请(ANDA)或生物制品许可申请(BLA)的一部分获得批准/许可时,它就成为FDA批准的已批准产品的分析程序。该分析程序可由FDA认可的来源进行适用性验证,例如美国药典/国家处方集(USP/NF)的规范程序,或由公司作为NDA、ANDA或BLA提交文件的一部分提交并经FDA认可的有效方法。AMV实验和结果在AMV报告中进行了总结。AMV报告完成后,将起草一份SOP并转交给质量控制实验室。分析方法的有效性是分析方法和产品开发生命周期中的关键成就。
2、方法验证的类型
一个完善的分析程序应经过各种类型和级别的验证,以证明方法的可靠性、适用性和目的。根据产品开发阶段和方法的目的,方法验证分为三类:完全验证、部分验证和交叉验证(full validation, partial validation, and cross-validation)。
对于新开发和实施的方法,或者对现有方法的更改对关键成分或程序范围有影响时,需要进行全面验证。
部分验证评估对已验证方法的更改和修改。更改或修改可能涉及检测系统、样品处理、定量范围或基质的组成。部分验证的范围可以从确定日内准确性和精度的范围,到根据更改对方法的可靠性和一致性的影响进行近乎全面的验证。属于部分方法验证的修改或更改包括但不限于以下内容:
• 实验室之间的生物分析方法转移
• 分析方法的变化(例如检测系统的变化)
• 样本处理程序的变化
• 样本量变化(例如,儿科样本量减少)
• 仪器和/或软件平台的变化
• 检测范围的扩展
• 采集生物液体时抗凝剂(而非反离子)的变化(例如从肝素变为EDTA
• 物种内基质的改变(例如从人血浆改为人血)或基质内物种的改变(例如从大鼠血浆改为小鼠血浆)
• 基质变化(例如脑脊液)
• 证明分析物在同时服用药物时的选择性
• 关键试剂的变化(例如批次间变化、试剂变化)
交叉验证用于比较两种或多种生物分析方法的验证参数,以在同一项研究或不同研究中生成数据。例如,当多个合同分析研究实验室参与分析多站点临床试验的样本时,可以执行方法的交叉验证,以确保方法的适用性和实验室间的可靠性。
3、分析方法验证
药典方法,例如已出版的专著、USP/NF、国际官方分析协作协会的官方分析方法、药典(USP和Ph. Eur.)的一般章节或其他公认的标准参考资料,均经过监管机构验证并得到认可。在使用前无需进行全面验证。但是,这些方法需要验证在实际使用条件下的适用性,以确定产品在药典测试中的基质效应。方法适用性或方法验证报告用于记录验证适用性的结果。标准测试(如pH值或灼烧残渣测试)无需进行方法适用性验证。
4、分析方法转移和分析方法可比性
有关分析方法转移(Analytical Method Transfer , AMT)和分析方法可比性(Analytical Method Comparability, AMC)的详细指导,请参阅美国食品和药物管理局(USFDA)的《行业指导:药物和生物制品的分析程序和方法验证》。美国药典(USP)<1224>为可接受的AMT替代策略提供了指导。PDA技术报告第57号:《生物制品的分析方法验证和转移》也提供了有关AMT和AMC的详细指导。
在分析方法和产品开发周期内,随着对工艺的认知加深或新实验室的需求出现,可能会出现方法转移、修改或替换。将分析方法转移至新实验室或实施修改或替代方法的基本实验是相似的。
AMC或AMT的范围在很大程度上取决于产品和方法在开发生命周期中的阶段。例如,在产品开发或毒理学研究阶段,对产品和工艺的整体了解较少,而方法在后期阶段(如Phase 2)是合格的,并且可以获得更大的方法性能信息数据库。因此,在后期阶段开展AMT/AMC研究的风险评估策略可能相当于部分验证,其范围从日内准确性和精度的确定到近乎全面的验证。
4、全球供应链的技术转让
世界卫生组织于2011年发布了附件7指南,其中详细介绍了制药商在药品制造方面的技术转让。虽然世卫组织为在全球范围内销售药品的制造商制定了指导方针,但该组织也参考了ICH Q8、Q9和Q10指南。该指南为全球制造商提供了基础,这些制造商向世界各地具有不同监管要求的国家供应活性药物成分和成品药物。
5、ICH Q14概述:分析程序开发
ICH大会于2018年6月批准了分析程序开发。该指南旨在协调和修订ICH Q2(R1)《分析程序验证指南》中提出的科学方法,同时改善行业与监管机构之间的沟通,以优化审查流程,并采用更健全、基于风险的方法进行上市后变更管理。ICH Q2(R1)中描述的原则将得到更新,以提供一个涵盖Q6A和Q6B范围内产品的框架。更新后的指南还将补充ICH Q8至Q13中的概念。
目前,该指南刚刚完成公众咨询流程,公众意见将提交给ICH,供流程的第三步参考。许多审查员评论说,该指南没有提供改进方法开发方法所建议的好处。其他评论指出Q14是Q2的延伸,但包含了对分析方法开发的生命周期管理方法。有几条评论涉及实时发布测试的方法,该方法在Q6A和Q6B中进行了讨论,并将这些概念纳入Q14。许多审查员质疑,遵循Q14是否允许制造商在批准后对分析测试方法进行更改时拥有更大的灵活性。总体而言,该指南草案令人困惑且范围有限。
ICH Q14将在明年继续发展,包括在2023年5月实施Q2和Q14工作组时采用步骤4。与此同时,制造商可以继续遵循ICH Q2中描述的分析方法开发概念。
6、其他相关指南概览
关于将“分析质量源于设计”(Analytical Quality by Design,AQbD)原则应用于药品药典标准的意见征询期已于2019年8月结束。英国药品和保健产品管理局(MHRA)希望通过这一过程实现其两大首要目标:通过强化系统确保药品安全生产,以及支持和促进创新。为了此次咨询,该机构利用实际案例研究来促进英国药典、良好生产与分销规范(GMDP)监察局和行业专家之间的合作。这种方法包括将“设计质量分析”(AQbd)应用于阿托伐他汀片剂的分析方法研究。制造商捐赠了测定程序,以协助调查。通过这种合作,同行们能够使用ATP定义方法性能,并评估与药典方法的相关性。这个过程有助于从实际的角度定义方法性能参数。这种定义方法性能的合作过程是监管机构与行业领导全球制药创新努力的一个例子。
结论
分析开发是药品和生物产品开发中一个持续的过程。既往以来,新开发的方法必须进行质量、真实性和有效性(quality, validity, and effectiveness)评估,即使是过去测试过的方法也必须重新检查,以确保其适用于产品。随着知识从过去的经验和方法中建立起来,不断发展的法规和指南塑造了分析开发过程的发展。
参考文献:
1. United States Pharmacopeia and National Formulary (USP 41-NF 36). <1220> Analytical procedure life cycle [draft chapter]. Presented at United States Pharmacopeial Convention (USP); 2016. https://www. uspnf.com/sites/default/files/usp_pdf/EN/USPNF/usp-nf-notices/gc-1220-pre-post-20210924.pdf
2. Smith MJ, et al. Technical Report No. 57-2: Analytical method development and qualification for biotechnology products. Parenteral Drug Association; 2015.
3. International Council for Harmonisation. Validation of analytical procedures: Text and methodology Q2(R1). Step 4 version. Dated November 2005. https://database.ich.org/sites/default/files/Q2%28R1%29%20Guideline.pdf
4. Namjoshi S, et al. Quality by design: Development of the quality target product profile (QTPP) for semisolid topical products. Pharmaceutics. Published 23 March 2020. https://pubmed.ncbi.nlm.nih.gov/32210126/
5. Jackson P, et al. Using the analytical target profile to drive the analytical method lifecycle. Anal Chem. Published 9 January 2019. https://pubs. acs.org/doi/10.1021/acs.analchem.8b04596
6. ter Horst, JP, et al. Implementation of quality by design (QbD) principles in regulatory dossiers of medicinal products in the European Union (EU) Between 2014 and 2019. Ther Innov Regul Sci. Published 13 January 2021. https://doi.org/10.1007/s43441-020-00254-9
7. European Medicines Agency. Quality by design. http://www.ema. europa.eu/ema/index.jsp?curl=pages/regulation/document_listing/ document_listing_000162.jsp.
8. Yu LX, et al. Understanding pharmaceutical quality by design. AAPS J. Published online 23 May 2014. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4070262/
9. International Council for Harmonisation. Pharmaceutical Development Q8(R2). Step 4 version. Dated August 2009. https:// database.ich.org/sites/default/files/Q8_R2_Guideline.pdf
10. Environmental Protection Agency. Selection and application of an analytical method. In: Multi-agency Radiological Laboratory Analytical Protocols Manual (MARLAP). Dated July 2004. https://www.epa.gov/sites/default/files/2015-05/documents/402-b-04-001a-06-final.pdf
11. Tome T, et al. Development and optimization of liquid chromatography analytical methods by using AQbD principles: Overview and recent advances. Org Process Res Dev. Published 14 August 2019. https://pubs.acs.org/doi/10.1021/acs.oprd.9b00238
12. Food and Drug Administration. Analytical procedures and methods validation for drugs and biologics [guidance]. Current as of 20 April 2020. https://www.federalregister.gov/ documents/2015/07/27/2015-18270/analytical-procedures-andmethods-validation-for-drugs-and-biologics-guidance-for-industry
13. Food and Drug Administration. Bioanalytical method validation guidance for industry [guidance]. Current as of 29 April 2020. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioanalytical-method-validation-guidance-industry
14. United States Pharmacopeia and National Formulary (USP 41-NF 36). <1225> Validation of compendial procedures. United States Pharmacopeial Convention (USP); 2016. https://www.ofnisystems. com/wp-content/uploads/2013/12/USP36_1225.pdf
15. United States Pharmacopeia and National Formulary (USP 41-NF 36). <1226> Verification of compendial procedures. United States Pharmacopeial Convention (USP); 2016. https://file.wuxuwang.com/yaopinbz/USP36-NF31/USP36-NF31_01_240.pdf
16. United States Pharmacopeia and National Formulary (USP 41-NF 36). <1058> Analytical Instrument Qualification. United States Pharmacopeial Convention (USP); 2016. https://www.drugfuture. com/Pharmacopoeia/usp35/PDF/0594-0598%20%5B1058%5D%20 ANALYTICAL%20INSTRUMENT%20QUALIFICATION.pdf
17. Food and Drug Administration. Process validation: General principles and practices [guidance]. Current as of 24 August 2018. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/process-validation-general-principles-and-practices
18. European Medicines Agency. Process validation for finished products- Information and data to be provided in regulatory submissions –Scientific guideline. Last updated 21 November 2016. https://www.ema.europa.eu/en/process-validation-finished-products-informationdata-be-provided-regulatory-submissions-scientific
19. International Council for Harmonisation. M4: The Common Technical Document (CTD) M4Q: Quality. https://www.ich.org/ page/ctd
20. International Council for Harmonisation. Quality guidelines. https:// www.ich.org/page/quality-guidelines
21. European Directorate for the Quality of Medicines and Healthcare. Quality management documents: Validation/verification of analytical procedures. Revision in force July 2020. https://www.edqm.eu/en/quality-management-qm-documents
22. European Medicines Agency. Bioanalytical method validation guidance for industry. Dated May 2018. https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf
23. Organization for Economic Co-operation and Development.Guidance document for single laboratory validation of quantitative analytical method, No. 204. Dated 14 July 2014. https://www.oecd.org/chemicalsafety/testing/series-testing-assessment-publicationsnumber.htm
24. Briggs RJ, et al. Method transfer, partial validation, and cross validation: Recommendations for best practices and harmonization from the global bioanalysis consortium harmonization team. AAPS J. Published online 5 September 2014. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4389741/#:~:text=Partial%20validations%20are%20 performed%20to,full%20validation%20would%20be%20required
25. United States Pharmacopeia and National Formulary (USP 41- NF 36). <1224> Analytical instrument qualification. United States Pharmacopeial Convention (USP); 2016.
26. World Health Organization. WHO guidelines on transfer of technology in pharmaceutical manufacturing. Dated 2011. https:// extranet.who.int/pqweb/sites/default/files/documents/TRS_961_Annex7_2011.pdf
27. Eglovitch, J. ICH Q14: Pharma groups want clarity on benefits of enhanced approach. Regulatory Focus. Published 4 October 2022. https://www.raps.org/news-and-articles/news-articles/2022/10/ichq14-pharma-groups-want-clarity-on-benefits-of
28. Medicines and Healthcare products Regulatory Agency. Consultation on the application of analytical quality by design concepts to pharmacopeial standards for medicines. Last updated 12 August 2020. https://www.gov.uk/government/consultations/consultation-onthe-application-of-analytical-quality-by-design-aqbd-principles-topharmacopoeial- standards-for-medicines

来源:同写意


