您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2020-05-26 15:14
一次性使用内镜用活体取样钳通过手柄操作传递、控制头部工作,通过内镜通道(如消化道内镜、呼吸道内镜等)完成活组织取样。
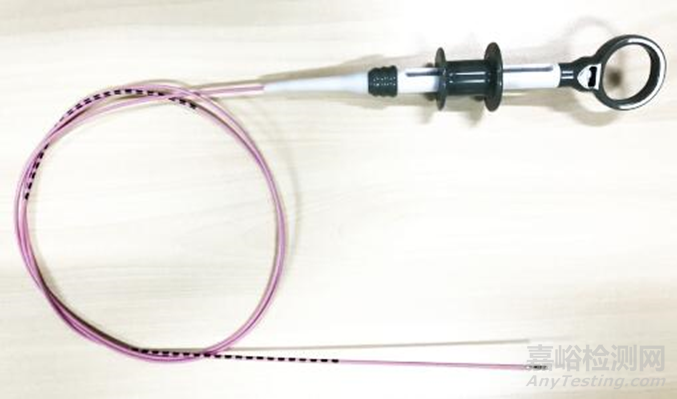
图1 典型一次性使用内镜用活体取样钳(带包塑层)

图2钳头形式分别为带窗平口型、鳄口型、带针型
一次性使用内镜用活体取样钳的产品主要危险(源)
|
危险(源)的分类 |
危险(源)的形成因素 |
可能的后果 |
|---|---|---|
|
生物学危险(源)
|
生产环境控制不好;产品清洁度不好;灭菌操作不严格;包装破损;使用时操作不规范。 |
产品带菌,引起患者感染。小分子物质残留量过大,造成毒性危害。 |
|
原材料控制不严;生产工艺控制不严;后处理未达到要求。 |
造成毒性危害;生物相容性不符合要求。 |
|
|
使用不当、标识不清。 |
引起感染、交叉感染。 |
|
|
未按照工艺要求配料;添加剂或助剂使用比例不正确。 |
生物相容性不符合要求。 |
|
|
环境危险(源) |
储运条件(如温度、湿度)不符合要求。 |
产品老化;无菌有效期缩短。 |
|
储运、使用过程中发生意外的机械性破坏。 |
产品使用性能无法得到保证。 |
|
|
使用后的产品没有按照要求集中销毁。 |
造成环境污染或交叉感染。 |
|
|
与医疗器械使用有关的危险(源) |
标记不清晰、错误;没有按照要求进行标记 |
错误使用;储存错误;产品辨别错误。 |
|
包装破损无法识别;操作要点不突出;不适当的操作说明,如: (1)和医疗器械一起使用的附件规范不适当 (2)预先检查规范不适当 (3)操作说明书过于复杂 (4)服务规范不适当 |
无法保证使用安全性;导致操作失误。 |
|
|
操作不熟练、操作失误;取样过大,无法取出,或出血过多。 |
样本掉落,肿瘤细胞种植转移,出血过多。 |
|
|
取样钳类型选用错误。 |
取样不满意。 |
|
|
对操作人员警示不足。 |
重复使用;二次灭菌;使用者出现过敏、刺激反应。 |
|
|
重复使用。 |
交叉感染。 |
|
|
不适当不合适或过于复杂的使用者接口 |
操作方法、注意事项、储存方法、警示事项等表述不清。 |
取样失败。 |
|
功能性失效和老化引起的危险(源) |
没有标识产品有效期。 |
超出有效期的产品被使用,造成细菌感染或因材料老化而导致产品性能不符合要求。 |
|
没有进行包装确认。 |
不能确保产品无菌,从而导致出现细菌感染。 |
|
|
产品标识没有明确。 |
出现细菌感染、交叉感染、以及粘膜损伤等现象。 |
|
|
钳头无法打开或闭合,控制开关失灵,断裂,钳头不锐利或咬切性能差。产品表面不光滑,有毛刺。 |
取样失败、样本掉落腔内。对内镜钳道有损伤,可能对患者有扎伤。 |
一次性使用内镜用活体取样钳的研发实验要求
应至少以下方面开展研究:
1.产品性能研究
应当提供产品性能研究资料以及产品技术要求的研究和编制说明,包括有效性、安全性指标以及与质量控制相关的其他指标的确定依据、所采用的标准或方法、采用的原因及理论基础等。对于首次应用于医疗器械的新材料,应提供该材料适用性相关研究资料。
2.生物相容性评价研究
产品首次注册时应根据产品所用材料及与人体的接触性质,按照GB/T16886.1-2011《医疗器械生物学评价第1部分:风险管理过程中的评价与试验》标准进行评价,若进行生物学评价试验,至少应进行细胞毒性、皮内反应、致敏的生物学评价研究。
3.灭菌/消毒工艺研究
(1)应明确灭菌工艺(方法和参数)和无菌保证水平(SAL),并提供灭菌确认报告。并对残留毒性提供研究报告。可根据适用情况,按照GB 18279.1-2015 《医疗保健产品灭菌环氧乙烷第1部分:医疗器械灭菌过程的开发、确认和常规控制的要求》、GB /T 18279.2-2015《医疗保健产品的灭菌环氧乙烷第2部分:GB 18279.1应用指南》、GB 18280.1-2015 《医疗保健产品灭菌辐射第1部分:医疗器械灭菌过程的开发、确认和常规控制要求》、GB 18280.2-2015 《医疗保健产品灭菌辐射第2部分:建立灭菌剂量》、GB/T 18280.3-2015 《医疗保健产品灭菌辐射第3部分:剂量测量指南》等标准的要求开展研究。
(2)若灭菌使用的方法容易出现残留,如环氧乙烷灭菌,应当明确残留物信息及采取的处理方法,并提供研究资料。
4.产品有效期和包装研究
产品有效期的验证可采用实时老化或加速老化的研究。实时老化的研究是唯一能够反映产品在规定储存条件下实际稳定性要求的方法,应遵循极限试验和过载试验原则。加速老化研究试验的具体要求可参照YY/T 0681.1-2009《无菌医疗器械包装试验方法第1部分:加速老化试验指南》系列标准。
对于包装的有效期验证,建议提交在选择恰当的材料和包装结构合格后的最终成品包装的初始完整性和维持完整性的检测结果。在进行加速老化试验研究时应注意:产品选择的环境条件的老化机制应与宣称的运输储存条件真实下发生产品老化的机制相匹配。对于在加速老化研究中可能导致产品变性而不适于选择加速老化试验方法研究其包装有效期验证的,应以实时老化方法测定和验证。
包装及包装完整性:在宣称的有效期内以及运输储存条件下,保持包装完整性的依据,可参考GB/T 19633.1-2015《最终灭菌医疗器械包装第1部分:材料、无菌屏障系统和包装系统的要求》、GB/T 19633.2-2015《最终灭菌医疗器械包装第2部分:成形、密封和装配过程的确认的要求》、YY/T 0698.1-2011《最终灭菌医疗器械包装材料第1部分:吸塑包装共挤塑料膜要求和试验方法》等系列标准提供研究资料。
5.其他研究
证明产品安全性、有效性的其他研究资料。该产品如包含镀层等,应对镀层的附着力、牢固性有相应的研究资料。申报资料中应明示与患者接触部分的材料,其中金属材料应标明牌号和(或)代号,并提供金属材料的化学成分试验报告(可以是由供货商提供的报告);高分子材料应明确材料的商品名或牌号(如有)。
一次性使用内镜用活体取样钳的主要性能指标
一次性使用内镜用活体取样钳的基本技术性能指标包括但不限于以下内容,申请人可根据产品自身特点,参考相应的国家、行业标准制定产品技术要求,如有不适用条款(包括国家标准、行业标准要求),申请人应在申报资料中说明理由。
1.外观
1.1软管外观、盘绕。
1.2钳头齿形。
1.3钳头外观。
2.尺寸
2.1插入部分最大外径。
2.2工作长度。
2.3钳头最大张开角度或幅度。
3.使用性能
3.1取样钳钳头开闭的要求。
3.2取样钳连接部位的要求。
3.3旋转性能(如适用)。
4.定位针的要求(如适用)。
5.钳头硬度。
6.钳头表面粗糙度。
7.耐腐蚀性能。
8.化学性能
根据不同材料的特性,申请人应对产品与人体接触部分的高分子材料的化学性能制定相应要求,如酸碱度、重金属、还原物质、蒸发残留物等。环氧乙烷灭菌的产品应规定环氧乙烷残留量不得大于10μg/g。
9.无菌要求。
10.镀层的要求(如适用)。
11.企业对宣称的所有其他技术参数和功能,均应在产品技术要求中予以规定。

来源:嘉峪检测网


