近日,美国FDA更新了“Requests for Feedback and Meetings for Medical Device Submissions: The Q-Submission Program”最终指导文件。
本指导文件的目的是概述提交者可用的机制,通过这些机制,他们可以申请以书面形式或在与美国食品药品监督管理局 (FDA)会议期间对以下潜在或计划中的内容请求反馈:
上市前批准 (PMA)申请
人道主义器械豁免 (HDE)
自动III类指定的评估(De Nove请求)
上市前通告(510(k))提交
临床实验室改进修正案(CLIA)豁免申请(CW)
Dual 510(k)和CLIA豁免(Duals)申请
附件分类请求
提交给生物制品评估与研究中心(CBER)的某些研究性新药申请(IND)
生物制剂许可申请(BLA)(特别是根据《公共卫生服务 (PHS) 法》第 351 条作为生物制品进行监管的器械 IND 和 BLA)
指导文件具体内容如下:
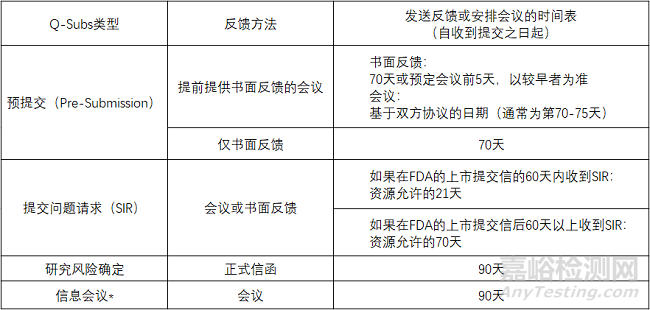
预提交(Pre-Subs)
预提交包括提交人对美国食品药品监督管理局反馈的正式书面请求,该请求以正式书面回复的形式提供。
如果提交人有需要,则以正式书面反馈的形式在会议后提供。会议期间发生的所有讨论都记录在会议纪要中,会议纪要由提交人起草并提交给FDA审查。
提交问题请求(SIRs)
SIRs是通过书面反馈或会议就解决上市提交保留函,CW保留函,IDE函或IND临床保留函中传达的问题的拟议方法请求FDA反馈。为了进一步澄清SIRs的范围,以下是本指南中适用的上市提交保留函:
510(k)s、De Nove请求、CWs和DUALs所需的其他信息
重大缺陷,不可批准,可批准缺陷,可批准待GMP,以及PMA和HDE的PAS条件批准
生物制剂许可证申请(BLAs)的完整回复函
SIR旨在促进FDA和提交者之间的互动,以快速解决这些信件中确定的问题,从而使项目能够向前推进,使提交者能够在正式回复中充分解决悬而未决的问题。SIR可用于讨论解决FDA信函中确定的问题的计划方法或策略。然而,SIR不应用于要求FDA预先审查预期的正式回应以评估充分性。
无论是否提交SIR,提交者都应在要求的时间表内对FDA收到的任何信件做出正式回应。
请注意,SIR不适合讨论传达最终决定的信件,例如实质上不等同,撤回和删除。
研究风险确定
研究风险确定是指要求FDA确定计划的医疗器械临床研究为显著风险(SR),非显著风险(NSR)还是适用免于IDE法规(21 CFR第812部分)定义的IDE法规。
对于不可豁免的研究,制造商负责进行初始风险确定(SR或NSR),并将其提交给机构审查委员会(IRB)。FDA为研究器械是SR还是NSR的最终仲裁者,并在IDE提交给FDA、运营商、临床研究者或IRB要求时做出决定。
信息会议
信息会议是要求与FDA分享信息,不需要反馈。这种信息共享有助于概述正在进行的器械开发的情况(特别是在未来6-12个月内计划多次提交的情况下),并使FDA审查小组熟悉新器械与当前可用器械在技术上的显着差异。虽然FDA工作人员可能会在信息会议上提出澄清问题,但他们通常会在会议期间倾听,且不提供任何反馈。
其他Q-Subs类型
除了上面列出的Q-Subs类型之外,Q-Subs还提供了一种机制来跟踪其他FDA程序指导文件中描述的相互作用。目前,除上述Q-Subs外,Q-Subs程序中跟踪的交互包括以下内容:
PMA第100天会议
FDA指南中描述的协议和决定会议
FDA指南中描述的与突破性器械计划相关的提交文件
FDA指南中描述的与更安全技术计划(“STeP”)相关的提交文件
FDA指南中描述的附件分类请求
这些其他Q-Subs类型的政策和程序可以在各自的指导文件中找到。
Q-Subs程序的其他用途
有些交互不符合上述Q-Subs类型的定义,并且尚未为其创建新的正式Q-Subs类型。当不存在新的Q-Subs类型来跟踪特定类型的交互时,FDA可以使用信息会议Q-Subs类型作为跟踪这些交互的工具。
一般来说,信息会议旨在提交人向FDA提供信息,而不需要FDA的反馈。但是,如果在尚未为该交互创建正式Q-Subs类型的情况下,将信息会议Q-Subs用于跟踪目的,则可以按照使用信息会议Q-Subs类型的程序的规定提供反馈。
不在Q-subs计划内的交互
在Q-Subs计划的范围之外,还有其他几种机制,行业可以通过这些机制从FDA获得反馈。有些需要或需要另一种形式的正式提交,而有些可以通过非正式互动来解决。
Q-Subs类型和相应的反馈机制和时间表
Q-Subs流程
提交内容
为确保正确登录并便于审查Q-Sub,Q-Sub申请书中应包含以下内容。
联系信息
Q-Sub类型
反馈方法
会议信息
为了从FDA获得有意义的反馈,应在Q-Sub内使FDA轻松识别以下内容:
目的。Q-Sub的总体目标包括与FDA互动结果的目标。
器械或产品描述。解释器械的功能、构成器械基础的基本科学概念以及器械的重要物理和性能特征。
建议的用途或预期用途指示。包括器械将诊断、治疗、预防、治愈或缓解的疾病或状况的描述,以及器械拟用于的患者人群的描述。
监管历史。列出与FDA之前有关主题器械的任何相关通信,包括但不限于与主题Q-Sub相关的任何营销提交,IDE,513(g)和/或Q-Sub申请号。
强烈建议使用CDRH上市前审查提交封面提交给CDRH或CBER,以便于正确登录并及时发送给适当的审查小组。
您必须根据FD&C法案第745(A)(b)节提交Q-Sub的电子副本。
如果您要向CDRH提交,我们建议通过CDRH客户协作门户(“CDRH Portal”)以电子方式提交eCopy提交包。
一旦通过CDRH门户提交,Q-Sub将由CDRH文件控制中心(DCC)接收。或者,提交包可以邮寄到CDRH DCC。CDRH DCC的当前邮寄地址在医疗器械提交的eCopy计划中提供。
对于受CBER监管的产品,我们建议通过FDA电子提交网关以电子方式提交提交包。或者,可以通过CBER提交电子邮件收件箱(最大150MB)提交,或者通过邮件发送到CBER DCC。
除了上述内容,该指导文件中还提供了预提交(Pre-Sub)验收清单、会议纪要示例等,帮助制造商更好地运用Q-Sub的功能。