您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-05-28 08:36
1、QbD的来历
质量源于设计 = quality by design = QbD:相关理念最早源于20世纪70年代Toyota为提高汽车质量而提出的创造性的概念,并经过在通信、航空等领域的发展逐渐形成。1985年,著名质量管理学家Julan博士在总结其质量管控经验和方法的基础上,提出质量源于设计,该理论同Deming的质量十四点原则和PDCA循环、Feigenbaum的全面质量管理、今井正明的持续改善等方法和理念共同奠定了现代质量管理的基础。
2、FDA中QbD的缘由
美国FDA提出QbD理念,是从许多的药物不良事件和致死事件开始的。(具体许多案例可以网上查。国外的药业不是一开始就好,死亡事件大把大把的,到今天这样的地步,也是利益与政治的不断平衡的结果。)
20世纪40年代初,为了应对大量的药物致死事件,美国FDA建立了GMP标准,而GMP控制质量的主要方式是质量源于检测,即QbT (quality by test);20世纪60年代初,为应对药物“反应停”导致1万多名婴儿畸形的事件,颁布了《Kefauver-Harris药品修正案》,从而保证药物的安全性和有效性;2002年,FDA发布了cGMP;2004年,FDA再次对cGMP进行了升级,发表了《Pharmaceutical cGMPs for the 21st century – A Risk Based Approach》,报告一文中正式提出了QbD的概念,并且被人用药品注册技术规定国际协调会议ICH纳入质量体系中。
ICH将QbD纳入到自己的质量体系当中后,用了多个章节来解释和阐述QbD的内涵和运用方法。ICHQ8对QbD的定义为:
译文
以合理的科学和质量风险管理为依据的,起始于预定的质量目标,注重对产品和工艺的理解以及对生产工艺过程控制的系统的研发方法。
原文
A systematic approach to development that begins with predefined objectives and emphasizes product and process understanding and process control, based on sound science and quality risk management.
3、ICH关于QbD的内容
ICH Q8至ICH Q11对QbD概念的理解与实施的简单介绍如下:
ICH Q8
详细介绍了在药品及其生产工艺的研发过程中如何应用科学策略和质量风险管理策略获取对产品和生产工艺的全面理解,从而为设计空间、质量标准和生产控制策略的制订提供科学依据,并为药品质量的风险管理提供坚实基础。
ICH Q9
有效的质量风险管理采用主动策略识别药物研发和生产阶段的潜在质量问题并加以控制,能够更高程度地保障药品的高质量,而且还有助于管理当局和申报企业均能更加科学、有效地进行风险决策。重点介绍了质量风险管理的原则和常用分析工具。其中,质量风险管理的两个重要原则为:一、风险评估应该基于科学并用于最终保护患者利益;二、风险管理的投入应该与风险等级对应,风险等级越高,管理投入应越大。
ICH Q10
详细阐述了如何采用QbD的理念、应用科学管理和质量风险管理这两大核心工具建立有效的药品质量系统。其中,科学管理依据已知的科学知识和在该药品生命周期内(药物研发、技术转移、商业化生产和产品停产)获得的对产品及其工艺的全面理解。药品质量系统的目标:获得符合目标产品质量属性要求的产品、建立并维持产品的受控状态以及促进产品质量的持续改进。药品质量系统的要素包括:工艺性能和产品质量的监控系统、校正和预防措施系统、变更管理系统以及工艺性能和产品质量的管理回顾。
ICH Q11
ICHQ11重点关注了原料药的开发和制造,制药公司在开发原料药过程中可以按照“传统”或“强化”的方法或联合两种方法进行。在传统的方法中,对工艺参数设定数据或者范围,原料药的控制策略通常是基于过程的重复性和测试,可以满足既定的验收标准。而强化方法更加广泛地使用风险管理以及科学知识来识别和理解影响关键质量属性(CQA)的工艺参数和单元操作,并在原料药整个生命周期中应用恰当的控制策略,其中也包括建立设计空间。
FDA认为,虽然QbD原理提出时间不长,但实际药品研发企业在药品质量控制过程中,已进行了很多QbD的实践,只是未曾上升到理论的高度。
所以,大家不必过于惊慌,或对QbD抱有一种高深莫测的态度。
4、QbD过程介绍
质量源于设计QbD,是一种系统的研发方法,其以预先设定目标为起始,基于可靠的科学和质量风险管理,强调对产品和生产过程的理解,及对工艺的控制,它是仿制药研发的有效工具。QbD理念是在仿制药研发上要求在对产品质量概况(QTPP)以及关键质量属性(CQAs)充分理解的基础上,对于关键工艺参数及其与CQAs间的关联以及潜在的高风险变量进行充分研究和筛选,并建立设计空间( DS),即影响产品CQAs的关键工艺参数范围组合,以此加强对制药过程的理解和控制,确保产品质量的持续控制。
一些关于QbD概念的英文缩写介绍
|
QTPP |
Quality Target Product Profile |
目标产品质量概况 |
|
CQA |
Critical Quality Attributes |
关键质量属性 |
|
CPP |
Critical Process Parameters |
关键工艺参数 |
|
QCPP |
Quality Critical Process Parameter |
质量关键工艺参数 |
|
RA |
Risk Assessment |
风险评估 |
|
DS |
Design Space |
设计空间 |
|
CS |
Control Strategy |
控制策略 |
|
PAR |
Proven Acceptable Range |
被证明可接受范围 |
|
CI |
Continual Improvement |
持续改进 |
QbD流程图
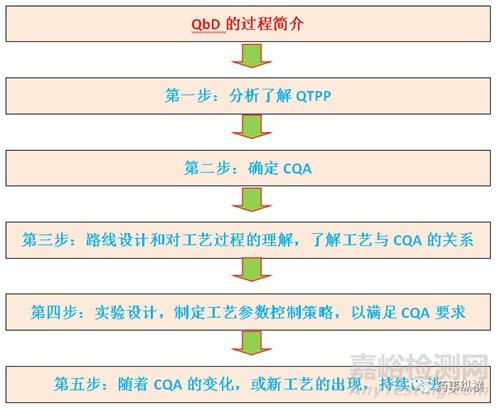
用中国的成语形容,我感觉就是“抽丝剥茧”。只不过将抽象的概念具体化了而已。
5、GSK具体案例
下面介绍一个具体的实例,来阐述QbD是如何运用于原料药开发过程中。
下图葛兰素史克公司开发的强效神经激肽受体拮抗剂--卡索匹坦甲磺酸盐。
合成路线如下:
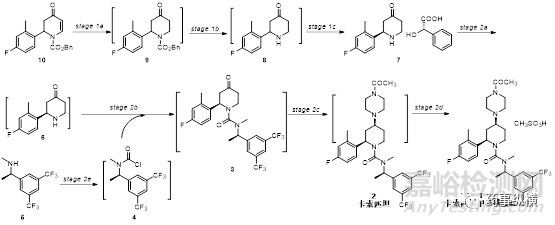
Stage 1过程中,二氢吡啶酮10经还原、水解、脱羧得到哌啶酮8。运用L-扁桃酸,经动态动力学拆分,得到R型哌啶酮7。
Stage2过程中,R型胺化合物5经与二氧化碳,氯化亚砜反应得到化合物4,与化合物6反应得到3。3与乙酰基哌嗪反应,并进一步还原得到2。加入甲基磺酸通过选择性析晶最终得到高纯度的化合物1--卡索匹坦甲基磺酸盐。
该合成路线较复杂,关注点较多,需要对每一步都进行详细的研究与探索。
下面以stage2a步骤为例,运用QbD原则进行的过程控制策略开发作介绍。
第一步:对原料药QTPP分析,确定CQA。
目前可以确定的CQA有原料药的立体异构体杂质。并且在stage 2a过程中,可能会伴随手性中心的消旋,因此需要对该步进行研究,以确保终产品异构体杂质合格可控。
第二步:对工艺过程的理解。
卡索匹坦有三个手性中心,理论上存在8个立体异构体。包括卡索匹坦及其异构体,以及另外6个非对映异构体,6个非对映异构体可以组成3组对映异构体。
这些异构体杂质的含量都是原料药的CQA,需要按照ICH的指导原则去控制。
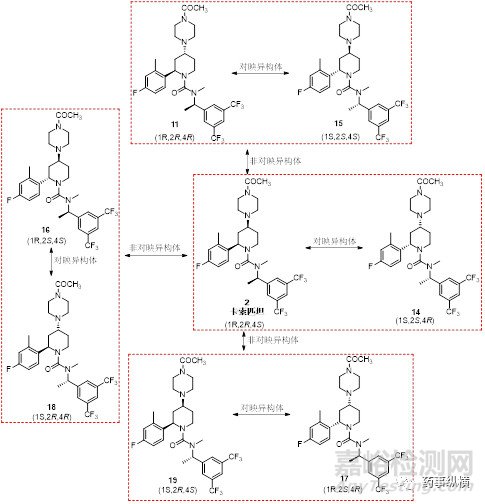
首先,对stage2a进行研究:
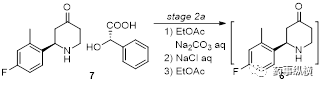
化合物7通过与碱中和生成化合物6。选用碳酸钠而不用碳酸氢钠,是因为碳酸钠水中溶解度更高,可以提高浓度。选用乙酸乙酯做溶剂是应为后续的步骤都用乙酸乙酯,为了统一。
考察化合物6的稳定性。
根据已有的知识基础,化合物6会存在如下的消旋化过程。化合物6的消旋化产物13会继续进行后续反应,以致最终产生卡索匹坦的异构体杂质,影响原料药CQA。
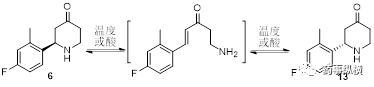
第三步:过程理解与控制策略。
对该步进行风险评估,推测反应温度和乙酸乙酯的量是潜在的质量关键工艺参数(QCPP)。
这两个参数在stage 2a的整个操作过程中都进行了考虑:
1) 化合物7悬浮于乙酸乙酯中;
2) 加入碳酸钠水溶液,搅拌直至全部溶解;
3) 两相分离;
4) 有机相分别用氯化钠水溶液和水洗;
5) 两相分离;
6) 有机相减压旋干。
首先研究发现,3-5项操作对原料药的CQA无影响。
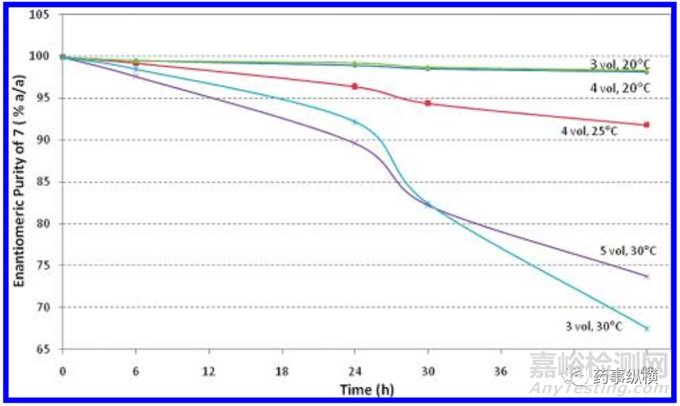
对操作1进行研究,尝试了5个实验,不同温度和不同乙酸乙酯体积下,随着时间的延长,化合物7的消旋情况。
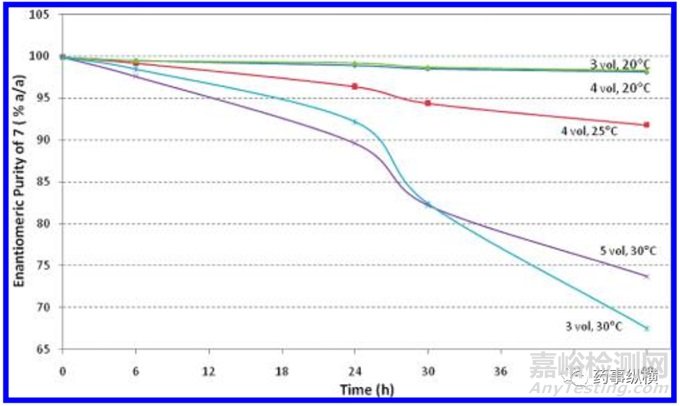
由图可知,化合物7的消旋化收温度和溶剂体积的影响。当为20摄氏度时,48小时以内基本不消旋,并且溶剂体积增大无影响。当温度升高至25摄氏度时,能够保证在6小时以内,原料的纯度都可以接受。所以该步设定温度不超过25摄氏度。
在工业生产时,滴加碳酸钠水溶液需要很长时间,可能会超过6小时,但研究发现,滴入碳酸钠水溶液后,化合物6很稳定,不会发生消旋作用。
对操作2进行研究,与操作1的研究基本相同,并且发现该步不影响原料药CQA。
对操作6进行研究,发现只有在减压蒸馏时,温度不超过25摄氏度,蒸馏终点乙酸乙酯含量大于2倍体积时,才能保证化合物6稳定,不消旋。
总结以上研究,对stage 2a的QCPPs和PARs总结如下:
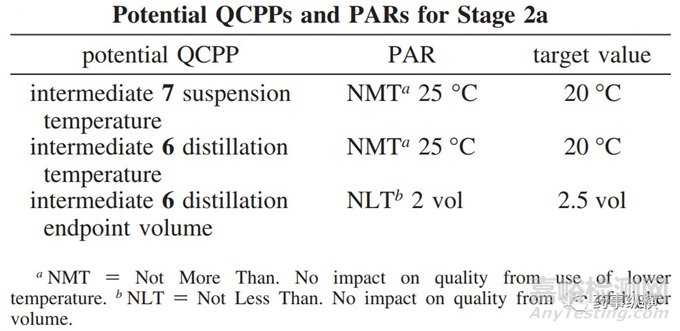
对stage2 按上述的范围操作,可确保原料药CQA里的手性异构体可以符合预定标准。
上述的操作可接受范围的确定只是实验室的小规模实验。
为了确保上述工艺范围,放大后也同样适用,尝试了在2L的设备上,模拟工艺生产设备和工业操作准则和原理,进行放大实验研究。并且也尝试了在可接受范围的极限值进行实验,研究发现卡索匹坦终产品质量同样合格。
结语
1,通过已有的知识对反应进行分析,建立工艺筛选模型。通过设计空间的知识对反应参数进行优化与研究,并最终获得可靠的工艺参数范围,对工艺参数范围的极限值研究,了解工艺的可靠性。
2,运用风险评估的知识,确定stage 2a的工艺参数范围,对原料药的CQA研究较小,不属于质量关键工艺参数(QCPP),只属于质量工艺参数(QPP)。
3,ICH关于QbD的指导文件,提供了一个系统结构化的方法来获取过程知识和制定可靠的工艺控制策略。
4,国内药企目前做的以仿制药为主,工艺路线一般较成熟,可以有许多文献参考资料,方向已经在那了,只需再细微的摸索一下最合适的工艺。并且许多仿制原料药已有相应的USP、EP等质量标准,大家对原料药的质量不是十分担心,倾向于买回来所有的杂质对照品,研究质量。对QbD的理念有运用,但不是很深入。不过这也正常,毕竟ICH原则原本是针对新药的。
5,随着国内的新药不断涌现,QbD的理念应该会越来越深入的被药企运用。与其等到那时再学,还不如现在就开始试着往自己的申报资料里,按QbD要求去研究,先学起来

来源:Internet


