您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-01-21 22:15
【导读】由于口服缓控释制剂在临床应用上具有一定的优越性,所以一直是国内新药研究的一个热点。如何控制其在体内确实具有缓释特征,进而保证其安全有效性,则是该剂型研发与评价的一个重点。从近几年所申报的此类品种分析,在口服缓控释制剂释放度的研究方面仍存在较多的问题。对此,提出关于口服缓控释制剂释放度研究的建议,供大家交流学习。
缓控释制剂相对于普通制剂而言,在安全、有效性方面具有一定的优点,例如可以延缓释放而减少给药次数,减小药物浓度波动从而降低毒副作用以及其他的治疗目的。近年来,随着制剂技术的不断发展及创新,对于缓控释制剂的研究越发深入,相应地缓控释制剂的申报量也呈现增加状态。
对于大多数口服缓控释制剂而言,相对于吸收行为,胃肠道的释放是主要的限速步骤,故对缓控释制剂进行体外释放度研究,无论是从前期产品的处方工艺研究以及最终产品的内在质量控制方面都具有重要意义。故在此,对于如何进行释放度的研究提出一些初步看法。
一、释放条件的选择
释放度系指一定剂型的药物在规定溶剂中释放的速度和程度。制剂的释放行为受自身因素和外界因素的影响。自身因素包括药物自身的特点(晶型、粒径、溶剂化物),辅料特点(种类、用量、质量),制剂生产过程(原辅料混合过程、制粒干燥过程、压片过程)等。外界因素系指释放度测定条件。释放条件的选择合适与否,密切关系到最终确定的质量标准能否切实控制产品质量。
释放度检查方法确定的总体原则是:首先释放条件应该具有一定区分能力,能够区分由于生产中关键参数改变(如控制释放行为的关键辅料的用量改变等)而可能影响到生物利用度的不同产品,但又不能过于敏感,以致于微小的变化均被视为不同。其次,所建立的方法应该比较稳定,能够准确客观地反映产品的释放情况。研究过程中,结合考虑各种外界条件对释放行为的影响,通常需对释放介质、转速、仪器进行详细地考察:
1.释放介质:释放介质的选择依赖于药物的理化性质(溶解性,稳定性、油水分配系数等)、生物学性质(吸收部位等)及口服后可能遇到的生理环境。在研究过程中,一般推荐选用水性介质,溶出介质的体积需使药物符合漏槽条件,通常选用500,900,1000ml。
由于不同pH下药物的溶解度、控制药物释放行为的关键辅料的水化、溶涨、溶蚀速度可能不同,故建议对不同pH(模拟胃肠道的生理环境,一般选用pH1-6.8的释放介质,有些情况下亦可考虑pH8.0的释放介质)条件下的释放行为进行考察。通常选择类似胃肠液的介质(pH1.2的人工胃液、pH4.5、6.8的缓冲液)或脱气后的新鲜蒸馏水。如药物的溶解性很差,也可在其中加入少量的表面活性剂,用量越少越好。根据以上研究结果,一则可以了解产品对口服后可能遇到的生理环境的敏感性,二则可以通过考察不同处方在以上释放介质中释放行为的差别,选择具有较强区分能力的条件。
为加强方法的区分能力,在保证药物在所选介质中的溶解行为符合漏槽条件的前提下,溶解度不宜过高,必要时需考虑离子强度和表面张力的影响。
2.转速:迄今,对于转速同胃肠道的运动尚未建立明确的关系。对于某些制剂而言,不同转速下的释放行为基本一致,说明产品释放特性受机械外力的影响较小。但对于大部分制剂而言,不同转速下的释放行为不同,例如溶蚀型制剂,转速越大,释放越快,故应考察产品在不同转速下的释放行为。转蓝法的常用转速为75-100rpm;桨法的常用转速为50-75rpm。转速过快,可能会导致对不同制剂释放行为的区分能力较差,所以不推荐首选过高转速。
3.仪器装置:对于仪器的选择,需考虑具体的剂型及可能的释药机制。通常建议选择药典中收载的仪器装置进行,一般情况下,片剂倾向于选择桨法,转篮法多用于胶囊及可能会漂浮的制剂。如采用其他特殊仪器,需提供充分的依据。
4.温度:常用释放介质温度为37.0±0.5℃。
在以上研究基础上建立的体外释放度检查方法,如未进行体内外相关性的验证,则只能做为控制产品质量的一种手段,不能预测产品体内的释药行为。建议研制单位能够进行一些体内外相关性的研究,一则可以进一步指导处方设计,二则可以优化体外释放条件,通过体外释放检查在一定程度上预测体内释药行为。
二、释放度检查方法的验证
释放度检查方法的验证主要包括三个方面:
1.释放条件和释放限度的合理性:对于释放条件的合理性,主要是评判所选定的条件是否可有效区分不同产品的质量,可以通过考察不同处方在不同释放条件下释放行为的差异进行验证,如能结合体内研究结果做进一步判定,则更具有说服力。对于释放限度的合理性,一般是要根据多批产品的检测结果以及体内研究结果确定。国外产品研究进程中,通常会对符合质量标准要求的实验室规模样品、临床样品和放大生产规模样品进行生物等效性研究,如结果显示生物等效,则在一定程度上对所确定的体外释放度检查方法的可控性进行了验证。
2.释放条件的耐受性验证,即需要验证释放条件的微小改变,是否会影响产品的释放行为。通常是根据实际操作中可能存在的误差,考察释放介质的体积(±1%)、释放介质的pH(±0.05)、温度(±0.5℃)以及转速(±5rpm)的微小变化对释放行为是否产生影响。如结果显示以上条件的微小变化对产品释放行为有较明显的影响,则说明所用条件的耐受性较差,不适宜于实际操作,应重新修订。
3.释放量测定方法的方法学验证,具体要求见方法学验证的相关指导原则。在这里需特别强调的是方法学验证过程中除常规考虑外,尚应关注:主药在释放介质中的稳定性;最佳取样量,以保证测定简便,尽量减小误差;滤器的性质,考证有效成分在滤器上是否有吸附。
三、释放特性研究
对于产品释放特性的研究,通常需绘制完整的释放曲线。为保证能绘制完整的释放曲线(包括上升曲线及达到平台的阶段),应选取足够多的取样测试点,通常前期取样点的间隔比较短,后期取样点间隔可相对延长。释放度考察时间要根据制剂释放时间长短不同而异,一般不宜短于给药间隔。
四、质量标准的建立
1、取样点的设置:通常质量标准中释放度检查需至少设置三个时间点,即我们所熟知的考察药物是否有突释现象,药物是否平稳释放,药物是否释放完全三个点。一般而言,第一点为1-2hr,药物释放量介于20-30%,第二点释放量约为50%,第三点为释放超过80%或药物释放已达到平台期的时间点。根据具体制剂不同的释药时间,可考虑适当增加释药测定点,以保证对产品的释药特性有比较全面的控制。例如USP25版中收载的普萘洛尔缓释胶囊释放度检查中设置了五个取样点:1.5h(不得超过30%)、4h(35-60%)、8h(55-80%)、14h(70-95%)、24h(80-110%)。对于某些产品,如在一特定时间段内的体外释放行为符合零级释放(例如从4-12小时内每小时释放5%),质量标准中除以上三个检测点外,尚可增订释药速率指标,即每小时的释放百分率。
2、释放度限度确定:释放度限度主要应根据临床用样品的平均检测结果进行确定。因为临床用样品一般为中试规模样品,其体外释放行为一则可以在很大程度上代表产品放大生产后的行为,二则其体外释放行为得到临床试验的验证,通过体内安全有效的结果可以支持体外释放限度的合理性。一般规定每个时间点上下浮动范围不得过 20%(即±10%)。某些情况下,如具有充分的理由,偏差浮动至25%以内也可接受;如超过25%的限度,则可能影响到产品的体内行为,建议进行生物等效性试验,验证上下限之间生物等效。
对于不同厂家研制的同一品种的缓控释制剂,如产品的释药机制不同,所建立的体外释放度测定方法可能不同,只要所建立的方法能切实控制产品质量即可,无需强求统一。USP25版中的缓释制剂基本都收载了几种不同的释放度测定方法,例如盐酸地尔硫卓缓释片(24h)项下列出了如下四种释放度检查方法:
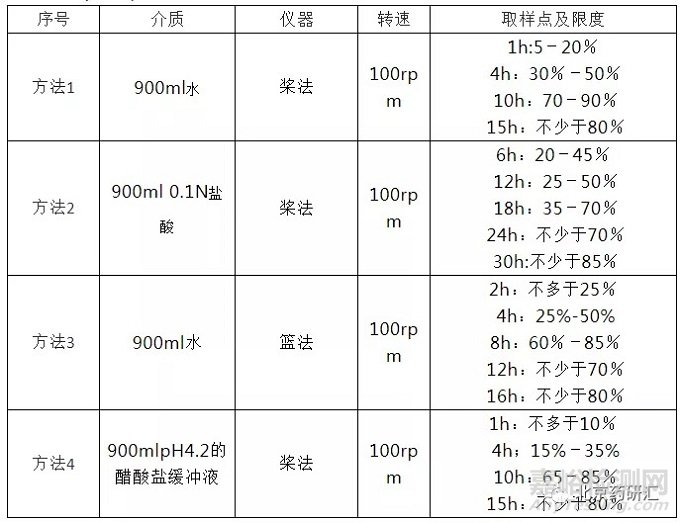
五、其他需关注的问题
释放度研究不仅可为最终确立质量标准提供重要技术依据,同时其研究结果也可为处方设计、生产控制及临床用药提供提示性信息。建议通过释放行为的研究,对释药机制做一定的探讨,一则便于了解控制药物释放行为的关键因素,在生产中重点控制,二则可以量化释放特性。
除此,尚应加强释放度研究中所揭示的相关信息的整合和认识。通过考察产品在不同条件下的释放行为,可以了解产品对口服后可能遇到的生理环境的敏感性。例如,某产品在酸性条件下溶出较好,对于正常患者而言,可以达到有效治疗血药浓度,可对于胃酸分泌较少的患者而言,血浓可能很低,根本无法达到有效治疗浓度,这就造成了明显的个体差异。如能有效重视释放度研究中揭露的信息,可为后续的临床个体用药提供参考依据。

来源:Internet


