您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-12-10 17:08
摘 要
最近几年CED发补最多的一条“有关物质:请参照国内外药典及工艺合成路线,对本品的有机杂质谱进一步分析和研究,并根据方法学验证结果,选择合适的有机杂质定量方式”制剂有机杂质谱(有关物质)该如何分析与控制是一个值得讨论的话题。
1名词解释
有机杂质:包括工艺中引入的有机杂质和降解产物等,可能是已知的或未知的、挥发性的或不挥发性的。
有关物质:化学结构与活性成分类似或具有渊源关系的有机有机杂质。
特定杂质:在新原料药规范(标准)中规定要检测并有特定的认可标准的有机杂质。它可以是已鉴定或未鉴定的有机杂质。—ICHQ3A
特定杂质:新药质量标准中的有机杂质检查项目应包括经研究和稳定性考察检出的,并在批量生产中出现的有机杂质和降解产物,并包括相应的限度。结构已知或未知的这类有机杂质属于特定有机杂质。—2015版《中国药典》通则9102
非特定杂质:在新原料药规范(标准)中,其限度在总认可标准中控制而不单独控制的有机杂质。
降解产物:由于放置时间过长,和(或)光照、温度、 pH 或水的作用,和(或)赋形剂和(或)直接接触容器密封系统互相反应而导致的药物分子发生化学变化而产生的新的分子(亦称分解产物)。
关于特定杂质与非特定杂质举例:以FLUPHENAZINE DIHYDROCHLORIDE为例,药典中收载了A-F六个有机杂质,impuritiesA、B、C、D为特定有机杂质,unspecifiedimpurities为非特定有机杂质,包含E、F。由此可知,该有关物质方法检测方法研究了A-F六个有机杂质,制定质量标准时,E、F作为非特定有机杂质(即未知有机杂质的控制方式)进行控制。

2有机杂质的产生及控制
1 原料引入的有机杂质
原料采购过程需索要工艺路线,质量标准及全检COA,检索各国药典的质量标准等,需研究有机杂质列表如下。
表1原料需研究有机杂质列表
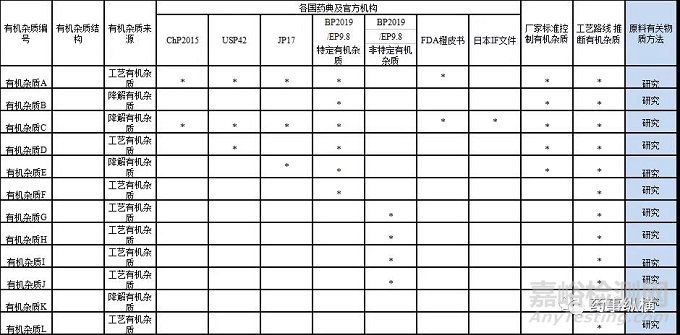
备注:*为各国药典收载或工艺推断出的有机杂质。
基于各国药典及合成工艺路线所推断的有机杂质,进行原料有关物质方法开发,结合原料的实际检测情况及制剂的相关要求,制定原料药有关物质的内控标准。
2 辅料引入有机杂质
辅料引入可分为三类
1类:辅料本身有吸收
如某些香精等,是可以被扣除的,不被认为是有机杂质峰。相关依据见下图:
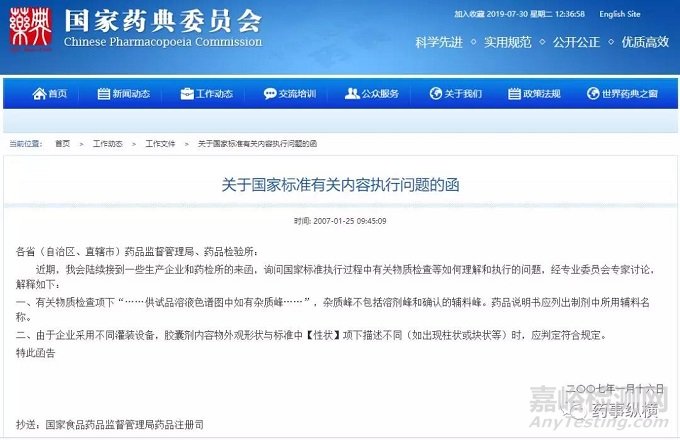
在制剂分析方法开发过程中如何避免辅料峰的干扰,可从以下几个方面考虑:
(1)避开辅料存在的吸收波长,使辅料无吸收或有较低吸收,以消除或降低辅料峰的干扰。
(2)根据辅料与主成分溶解性的差异,优化稀释剂使辅料不出峰。
(3)在不影响已知有机杂质分离的情况下,按未知有机杂质控制,此处理方法并非完全正确,仅供参考。
2类:辅料引入的有机杂质
该类有机杂质建议与辅料供应商联系,弄清该有机杂质产生的原因,若为生产过程引入的不明有机杂质,需更换辅料;若该有机杂质辅料厂家已做研究且有相关资料证明其使用范围内的安全性,无需更换辅料,将辅料厂家相关研究资料做为申报资料的附件即可。
3类:原辅料相容性引入的有机杂质
通常仿制药与原研处方是一致的,辅料本身与原料产生化学反应的概率较小(起码工艺过程反应的概率较小),但辅料加工过程的残留物质可能会与主成分相互作用产生有机杂质。举例如下,参照“原辅料相容性实验:还有很多你不知道的秘密—药事纵横”
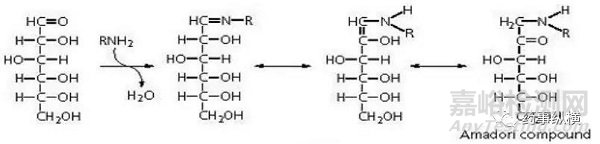
(1)如微晶纤维素含有蔗糖(还原糖)与伯胺和仲胺类药物会发生美拉德反应
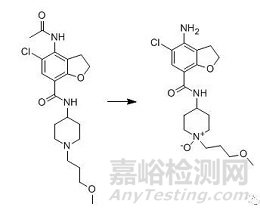
(2)羟丙基纤维素含有过氧化物与含叔胺基团的原料药反应
(3)某些API与硬脂酸镁中的镁离子产生化学反应。
(4)具有醇基或羧基的药物与某些辅料发生酯化反应
(5) 羧甲基淀粉钠生产过程中残留羟乙酸钠,会加速度罗西汀等的降解。
在进行原辅料相容性实验中,若有机杂质增长程度明显高于原研的影响因素测定结果,说明辅料不合格,建议更换辅料。
3 制剂工艺过程中引入的有机杂质
制剂工艺过程如片剂的湿法制粒、注射剂的酸碱调节等,使原料与辅料短时间处于某种特殊环境中,可能导致有机杂质的产生,该类有机杂质通常可通过以下两种方式进行研究。
(1)配合制剂日常检测,即可发现。
(2)通过适度的破坏实验,破坏强度大于工艺过程所接触的环境,但不可过度,具体的破坏实验应根据不同的剂型,不同的工艺而定,参照下表进行。
表2 破坏实验列表
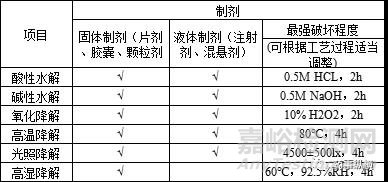
备注:“√”表示建议进行。
根据日常的样品检测结合适度的破坏实验,即可大致推断出可能产生的有机杂质,作为重点关注对象。
4 制剂稳定性放置过程中产生的有机杂质
通过影响因素、加速、长期稳定性等实验确定降解有机杂质,如下表所示
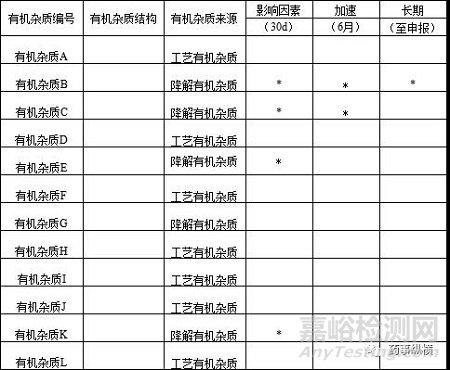
备注*表示降解出的有机杂质
3制剂有关物质研究及对方法的要求
参照2015版《中国药典》通则9102 药物有机杂质分析指导原则,除降解产物和毒性有机杂质外,在原料中已控制的有机杂质,在制剂中一般不再控制。截图如下:

通过上述内容,个人认为制剂有关物质应基于降解有机杂质和原料药的内控标准开展分析方法研究。关于原料引入的工艺有机杂质(明确的毒性有机杂质除外),制剂质量标准可以不定入,但是分析方法是否需具备工艺有机杂质的检出能力呢?
不同的人可能有不同的看法,我的个人看法:
(1)原料药的内控特定有机杂质,分析方法必须具备检测能力,理由为不同批原料,特定工艺有机杂质的检测量可能会存在差异,按未知有机杂质控制,制剂可能存在放行的风险。
(2)对于原料内控标准中的非特定有机杂质,通常原料内控标准中按未知有机杂质控制,无需研究。
表3 制剂需研究有机杂质列表
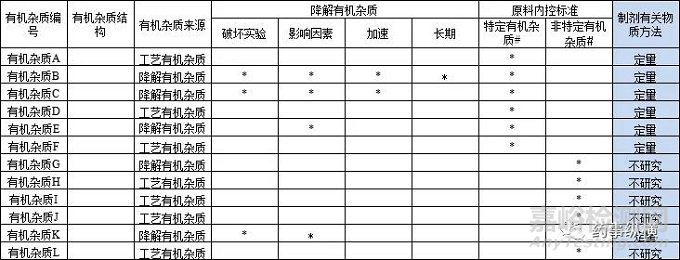
备注:*表示产生的有机杂质。#原料药内控特定有机杂质指标准中规定要检测并有特定的认可标准的有机杂质;原料药内控非特定有机杂质指标准中其限度在总认可标准中控制而不单独控制的有机杂质(即通过工艺路线推断及各国药典收载,但未按特定有机杂质收载至质量标准的一类有机杂质)。
4关于限度法控制有机杂质的一些思考
1用范围
适用于可不报告数据的有机杂质控制,如残留溶剂、基因毒性有机杂质等,通常在原料的起始物料、中间体有机杂质控制中常用,个人认为外购原料的内控标准也可以适当采用。
2 分离度该如何做?
建议主峰与临近峰分离度不得低于1.5,有机杂质间的分离度不得低于1.2。
若难度很大,基于风险评估,可适当放宽要求1、是否为降解有机杂质,若否,可适当放宽。理由为该类有机杂质不会增长且在质量标准中通常按未知有机杂质控制,分离度差,可按一个未知有机杂质的限度进行控制,即不影响产品的放行,也可确保产品的安全。2、若为降解有机杂质,则不建议放宽,因为存在稳定性样品检测无法放行的风险。
1 检测限该如何做
建议参照下述步骤进行[3]
(1)配制一个在限度浓度的有机杂质标准品溶液
(2)配制一个添加了在限度浓度的有机杂质标准品的样品标准品溶液
(3)配制了一个添加了(限度100%-RSD%*浓度)的有机杂质标准品的样品标准品溶液
*举例,若该类有机杂质的限度为0.05%,其RSD%通常为20%
如果1≤2且3<2,则差异可被检测出,该方法可以胜任有机杂质检测。
(4)质量标准中系统适用性增加各有机杂质灵敏度测试,参照如下程序进针:
空白溶液一针、检测限浓度(10%限度浓度)溶液一针、30%限度浓度溶液一针、限度浓度溶液一针,样品溶液一针。数据统计时,可报告**有机杂质小于限度的10%或小于限度的30%等。
致 谢
该篇文献感谢占小兵老师的指导,纠正了我一些错误的认识,也欢迎各位同仁批评指正,共同学习、提高。
参考文献
[1] 化学药品仿制药口服固体制剂质量和疗效一致性评价申报资料要求(试行)
[2] 化学药物(原料药和制剂)稳定性研究技术指导原则(修订)
[3] PF39(6)1200
[4] 药典委

来源:Internet


