您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2022-01-25 13:46
上海器审答疑汇总,有助于各位加深理解医疗器械方面相关细节知识。
一、胶体金产品需要变更内包装形式,是否必须申请登记事项变更注册?
根据《体外诊断试剂注册与备案管理办法》(总局令 第48号)第五章第七十八条要求,注册证载明的包装规格、产品技术要求、产品说明书等发生实质性变化,属于需要办理变更注册的事项,注册人应当向原注册部门申请办理变更注册手续;发生其他变化的,注册人应当按照质量管理体系要求做好相关工作,并在变化之日起30日内向原注册部门备案。
即注册证及其附件中若载明了产品内包装形式,当注册人变更产品内包装形式时,需要向原注册部门申请办理变更注册手续;注册证及其附件中若未载明该产品内包装形 作,并在变化之日起30日内向原注册部门备案。
二、体外诊断试剂有适用的国家标准品时,产品申报应关注哪些问题?
当体外诊断试剂有适用的国家标准品的,应当使用国家标准品对试剂进行检验。如有新的适用的国家标准品发布实施,延续注册的产品也应符合国家标准品的要求才能予以延续。
三、GB 9706.1-2020版已发布,在正式实施日期之前,企业可选择执行新版GB 9706.1吗?
根据《强制性国家标准管理办法》第三十九条的规定,“强制性国家标准发布后实施前,企业可以选择执行原强制性国家标准或者新强制性国家标准。新强制性国家标准实施后,原强制性国家标准同时废止。”企业可选择执行新版GB 9706.1标准。
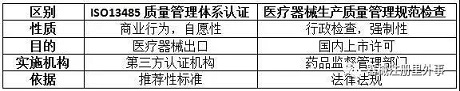
四、公司已经通过了ISO13485的认证和年度评估,办理医疗器械生产许可时药监局为什么还要来进行生产质量规范现场核查?
根据《医疗器械监督管理条例》规定,第二类、第三类医疗器械生产新开办、变更和延续均为行政许可事项,应当向所在地省、自治区、直辖市人民政府药品监督管理部门申请生产许可。《医疗器械生产质量管理规范》及附录是医疗器械生产企业必须执行的法规要求,是获得生产许可的基本条件。
ISO13485标准在国内等同转换为YY/T0287,是医疗器械质量管理体系推荐性行业标准,实施ISO13485认证的第三方认证机构不具备行政许可资格,通过第三方认证并不能作为上市许可的必要条件。
以下通过列表比较说明在我国ISO13485认证与《医疗器械生产质量管理规范》核查的区别:
五、质量部门独立行使权力在组织架构上应如何体现?有什么要求?
按照《医疗器械生产质量管理规范》的要求,医疗器械生产企业应建立质量管理部门,独立行使职能,对产品质量的相关事宜负有决策的权利。企业应成立专门的、独立的质量管理部门并由最高管理者或其授权代表直接管辖。质量管理部门应包括QA及QC的职能,具体职能应根据企业的实际情况在其职责文件中进行明确规定,例如质量管理、质量控制、质量检验、产品放行、让步接收、质量审核、质量数据统计分析、不合格品控制、内部审核、管理评审等。产品上市放行应由质量受权人负责,质量部门应对产品质量具有绝对的、独立的决策权,确保每批次原料、中间品、终产品符合质量要求。
六、同一注册单元的产品中包含试剂R1、R2、质控品、校准品,但是它们的使用期限各不相同,标签如何标示产品有效期?
同一注册单元中有多个组分,应以组分中最短的有效期/失效日期为该注册单元的有效期/失效日期,在标签上进行标注。
七、欲申请第二类医疗器械创新通道,想了解产品目前的状态是否可以进行申请第二类创新?
根据《上海市第二类创新医疗器械特别审查程序》,符合下列情形的本市申请注册的第二类医疗器械可以申请二类创新:
(一)产品主要工作原理或者作用机理为国内首创,产品性能或者安全性与同类产品比较有根本性改进,技术上处于国内领先水平,且具有显著的临床应用价值。
(二)申请人已完成产品的前期研究并具有基本定型产品,研究过程真实和受控,研究数据完整和可溯源。
(三)申请人通过其主导的技术创新活动,在中国依法拥有产品核心技术发明专利权,或者依法通过受让取得在中国发明专利权或其使用权,创新特别审查申请时间距专利授权公告日不超过5年;或者核心技术发明专利的申请已由国务院专利行政部门公开,并由国家知识产权局专利检索咨询中心出具检索报告,报告载明产品核心技术方案具备新颖性和创造性;或者产品技术国内领先,可填补本市该品种医疗器械的空白。

来源:上海器审


