您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2022-03-20 23:26
美国联邦公告Federal Register 于2022年2月23日发布:FDA提议修订21 CFR Part 820医疗器械质量体系法规QSR(Quality System Regulation),使其要求与国际上普遍共识的医疗器械质量管理体系标准ISO13485:2016版的要求更加趋于一致。修订后的法规将被称为QMSR (Quality Management System Regulation)。
1978年美国FDA首次发布医疗器械的CGMP(current Good Manufacturing Practice),并于1996年作出重大修订成为现行的QSR。1996年国际标准化组织也发布了第一版ISO13485标准。但自1996年后FDA未对医疗器械质量体系法规QSR做过重大修改。
FDA此次修订医疗器械质量体系法规的目的旨在推进医疗器械法规的全球协调一致,免除不必要的重复的医疗器械的法规要求和进入市场的障碍,并减轻企业负担。也是FDA为更好地实施FDA和其他四个国家共同参与的医疗器械单一审核程序Medical Device Single Audit Program (MDSAP)迈出的一大步。
当然FDA的新版QMSR和ISO13485:2016版还是有部分不同。例如,FDA对部分名词的定义与ISO13485不同;对于记录控制、标识和包装控制的要求也更严格,增加了唯一设备标识(UDI)的要求等等。征求意见稿的英文原文链接请点击文章左下方“阅读原文”。
对FDA新版法规的意见可以电子或纸质形式向FDA提交,提交电子版意见截止日期为2022年5月24日,可登录网站https://www.regulations.gov按照指引提交意见。提交纸质意见截止日期为2022年5月25日,邮寄至地址:Dockets Management Staff (HFA-305), Food and Drug Administration, 5630 Fishers Lane, Rm. 1061, Rockville, MD 20852.
FDA建议新版法规QMSR在正式公布一年后开始生效。
附:中文翻译如下
第 820 部分——质量管理体系法规QMSR
A 分部——一般规定
820.1 范围。
820.3 定义。
820.5 [预留的]
820.7 通过引用并入。
820.10 质量管理体系的要求。
820.15 澄清概念。
B 分部——补充规定
820.20-820.30 [预留的]
820.35 记录控制。
820.40 [预留的]
820.45 器械标记和包装控制。
C-O分部——[预留的]
授权
21 U.S.C. 351, 352, 360, 360c, 360d, 360e, 360h, 360i, 360j, 360l, 371,374, 381, 383; 42 U.S.C. 216, 262, 263a, 264.
A 分部——一般规定
§ 820.1 范围。
(a) 适用性。本质量管理体系法规 (QMSR) 中规定了现行的良好生产规范(CGMP) 要求。本部分的要求适用于所有供人类使用的成品医疗器械的设计、制造、包装、贴标签、存储、安装和维修所使用的方法以及所使用的设施和控制。本部分中的要求旨在确保成品医疗器械安全有效,并符合《联邦食品、药品和化妆品法》。任何从事成品器械设计、制造、包装、贴标签、存储、安装或维修的制造商都必须建立和维护适合其特定器械的质量管理体系。受本部分约束的制造商包括但不限于执行合同灭菌、安装、重新贴标签、再制造、再包装、或规格开发,以及执行这些功能的外国实体的初始分销商。如果制造商仅从事本部分要求的某些操作,而不从事其他操作,则该制造商只需遵守适用于其所从事操作的那些要求。
(1) 成品器械。本部分的规定应适用于,生产、进口或提供进口到美国任何州或领地、哥伦比亚特区或波多黎自治政体的,按本部分定义的任何供人类使用的成品器械。
(2) 组件或零件。本部分的规定不应用于成品器械的零件或部件的制造商,但鼓励此类制造商酌情考虑本法规的规定。
(3) 血液和血液成分。本部分的规定不适用于用于输血或进一步制造血液和血液成分的制造商。此类制造商受本章 F 小节的约束。
(4) HCT/Ps。本部分的规定应用于人体细胞、组织以及基于细胞和组织的产品(HCT/Ps) 的制造商,如本章 § 1271.3(d) 中所定义,这些器械( 须经上市前审查或通告,根据《联邦食品、药品和化妆品法》的器械条款提交申请或根据《公共卫生服务法》第 351 条提交生物产品许可申请,或豁免通告)。作为器械监管的 HCT/Ps 还需遵守本章第 1271 部分 C 小节中规定的供体资格要求以及本章第 1271 部分 D 小节中适用的现行良好组织实践要求。如果第 1271 部分的适用法规与本章其他部分的适用法规发生冲突,则具体适用于相关器械的法规应比一般的法规具有更高的优先级。
(b) 与《联邦食品、药品和化妆品法》下的其他要求相冲突。除另有明确说明外,本部分中器械的 QMSR 补充了本章其他部分的规定。如果本部分适用法规与本章其他部分的适用法规发生冲突,则具体适用于相关器械的法规应在其冲突范围内比更普遍适用的法规具有更高的优先级。此外,如果 ISO 13485的任何条款(通过引用并入,请参阅 § 820.7)与《联邦食品、药品和化妆品法》和/或其其他实施条例相冲突,则以《联邦食品、药品和化妆品法》的任何规定法案和/或其其他实施条例为准。
(c) 外国制造商。如果向美国提供器械进口的任何工厂、仓库或机构的所有者、经营者或代理人延迟、拒绝或限制检查,或拒绝允许进入或检查外国场地以确定符合本部分的目的,或用于制造、包装、储存、安装、加工或存放在此类工厂、仓库或机构中的用于进口的方法和设施和控制美国不符合《联邦食品、药品和化妆品法》第 520(f) 节和本部分的要求,则在该场地制造的器械根据《联邦食品、药品和化妆品法》第 501(h) 或 (j) 节被认为是假货,将根据《联邦食品、药品和化妆品法》第 801(a) 条被拒绝进入美国。
(d) 豁免或变通。 (1) 受《联邦食品、药品和化妆品法》第 520(f)(1) 条规定的任何要求(包括本部分规定的任何要求)约束的制造商可以根据《联邦食品、药品和化妆品法》第 520(f)(2) 条豁免或变更。豁免或变更申请应按照本章第 10.30 节规定的程序提交。
(2) 当 FDA 确定此类变通符合公众健康的最佳利益时,FDA 可以启动和批准本部分中的任何要求的变通。仅当该器械仍然存在公共卫生需求时,并且如果没有变通,该器械就会出现短缺,此类变通才会保持有效。
§ 820.3 定义。
除本节(b) 段中规定的外,ISO 13485 中的定义(以引用方式并入,参见 § 820.7)适用于本部分,并且不影响本标题中定义的类似术语的含义。
(a) 就本部分而言,下列术语是必要的,但未出现在 ISO 13485 中:
元件是指任何原材料、物质、部件、零件、软件、固件、标签或组件,旨在作为已完成、已包装并已贴标签的器械的一部分。
客户是指可以或确实收到该个人或组织预期或要求的产品或服务的个人或组织,包括用户。客户可以在组织内部或外部。
设计确认是指通过客观证据确定器械规格符合用户需求和预期用途。
联邦食品、药品和化妆品法是指《联邦食品、药品和化妆品法》,21 U.S.C. 321等等,经修订。
成品器械是指适合使用或能够运行的任何器械或任何器械的附件,无论其是否经过包装、贴标签或消毒。
作为器械监管的人体细胞、组织或基于细胞或组织的产品(HCT/P) 是指本章第1271.3(d) 节定义的不符合第 1271.10(a) 节标准的 HCT/P 章节的定义的器械,也被规定为一种医疗器械。
不合格是指不满足特定要求。
加工助剂是指在制造过程中使用或用于促进制造过程的任何材料或物质、在制造过程中产生的伴随成分或副产品成分,其作为残留物或杂质存在于成品器械中而并非制造商的设计或意图。
过程确认是指建立客观证据确定过程始终如一地产生满足其预定规格的结果或产品。
再制造商是指对成品器械进行加工、调节、翻新、重新包装、修复或执行任何其他行为,从而显着改变成品器械的性能或安全规格或预期用途的任何人。
返工是指对不合格产品采取的措施,使其在放行以供分销之前满足指定的要求。
最高管理层是指有权制定或更改制造商质量政策和质量管理体系的制造商的高层员工。
验证是指通过检查和提供客观证据来确认特定要求已得到满足。
(b) 《联邦食品、药品和化妆品法》第 201 节中的所有定义应适用于本部分下的质量管理体系规定,并应取代 ISO 13485 中的相关术语和定义(例如,器械和标记的定义)《联邦食品、药品和化妆品法》第 201(h) 和 (m) 节中适用于本部分并取代 ISO 13485(标记和医疗器械)中相关术语的定义。此外,以下术语和定义取代了 ISO 13485 中的相关术语和定义:
制造商是指设计、制造、生产、组装或加工成品器械的任何人。制造商包括但不限于执行合同灭菌、安装、重新贴标签、再制造、重新包装或规范开发等功能的人员,以及执行这些功能的外国实体的初始分销商。
产品是指元件、加工代理、加工中的器械、成品器械和退回的器械。
§ 820.5 [预留的]
§ 820.7 通过引用并入。
经联邦公报批准,某些资料通过引用并入本部分 5 U.S.C. 552(a) 和 1 CFR第51部分。所有批准的资料都可以在食品和药物管理局, Dockets Management Staff, 5630 Fishers Lane, Rm。1061, Rockville, MD 20852, 240-402-7500,和在国家档案和记录管理局 (NARA) 进行检查。有关在NARA 获得此资料的信息,请致电 202-741-6030,发送电子邮件fr.inspection@nara.gov,或从 www.archives.gov/ federal-register/ cfr/ ibr-locations.html。它可从以下来源获得:
(a) 国际标准化组织 (ISO), BIBC II, Chemin de Blandonnet 8, CP 401, 1214 Vernier,Geneva, Switzerland;+41-22-749-01-11;customerservice@iso.org, https://www.iso.org/ store.html.
(1)ISO13485, “Medical devices—Quality management systems—Requirements for regulatory purposes,” ISO 13485,“医疗器械—质量管理体系—用于法规的要求”,第三版,2016 年 3 月;IBR 批准 §§ 820.1;820.3;820.10;820.15;820.35;820.45。
(2) [预留的]
(b) [预留的]
§ 820.10 质量管理体系的要求。
如本部分§ 820.1(a) 所述的制造商必须:
(a) 文件化。文件化符合 ISO 13485(以引用方式并入,参见 § 820.7)和本部分要求的质量管理体系;和
(b) 适用的法规要求。遵守本标题中的其他适用法规要求,包括但不限于以下内容,以完全遵守列出的 ISO 13485 条款:
(1) 对于 ISO 13485 中的第 7.5.8 条,标识,制造商必须文件化一个系统,以根据第 830 部分的要求为医疗器械分配唯一的器械标识 UDI。
(2) 对于 ISO 13485 中的第 7.5.9.1 条,可追溯性—一般要求,制造商必须根据第 821 部分的要求形成可追溯性程序(如果适用)。
(3) 对于 ISO 13485 中的第 8.2.3 条,向监管机构报告,制造商必须将符合本章第 803 部分报告标准的投诉报告 FDA。
(4) 7.2.3、8.2.3、8.3.3 条的忠告性通知应按照 806 部分的要求处理。
(c) 设计和开发。II 类、III 类和下列 I 类器械的制造商必须符合 ISO 13485 中设计和开发第 7.3 条及其子条款的要求。I 类器械如下:
(1) 计算机软件自动化器械;和
(2) 下表所列器械:
表 1 为段落(c)(2)
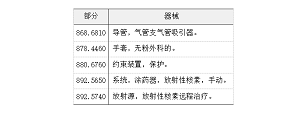
(d) 支持或维持生命的器械。支持或维持生命的器械,是指如果按照标记中提供的使用说明正确使用失效时预期可能会导致重大伤害的器械,的制造商必须遵守ISO 13485 中的第 7.5.9.2 条对于植入式器械追溯性中的要求,以及本部分中的所有其他适用要求。
(e) 执法。如果不遵守本部分中的任何适用要求,则根据《联邦食品、药品和化妆品法》第 501(h) 节的规定,器械被判断为掺假。此类器械以及对不遵守规定负责的任何人都将受到监管的约束。
§ 820.15 澄清概念。
制造商应将 ISO 13485(以引用方式并入,参见 § 820.7)中的以下术语理解如下:
(a) 组织应具有本部分定义的“制造商”的含义。
(b) 安全和性能,就本部分而言,应具有“安全性和有效性”的含义。“安全和性能”一词并不能免除制造商实施控制或其他措施以合理保证安全和有效性的义务。
(c) 过程确认应具有本部分定义的“过程确认”的含义。
B 分部——补充规定
§ 820.20-§ 820.30 [预留的]
§ 820.35 记录控制。
除了 ISO 13485 中第 4.2.5 条的要求(通过引用并入,参见 § 820.7),记录控制,制造商必须获得每个批准或重新批准记录的个人的签名,以及批准签署日期,在该记录中,并在某些记录中包括以下信息:
(a) 投诉记录。除了 ISO 13485 中的第 8.2.2 条,投诉处理,外制造商必须至少记录以下信息,用于根据 本章803部分必须向 FDA 报告的投诉,制造商确定必须调查的投诉,以及制造商不顾这些要求而调查的投诉:
(1) 器械名称;
(2) 收到投诉的日期;
(3) 任何唯一器械标识符 (UDI) 或通用产品代码 (UPC),以及任何其他器械的标识;
(4) 投诉人的姓名、地址、电话;
(5) 投诉的性质和内容;
(6) 采取的任何纠正措施;和
(7) 对投诉人的任何答复。
(b) 服务活动记录。为遵守 ISO 13485 服务活动中的第 7.5.4 条,制造商必须至少记录以下关于服务活动的信息:
(1) 服务的器械名称;
(2) 任何唯一器械标识符 (UDI) 或通用产品代码 (UPC),以及任何其他器械的标识;
(3) 服务日期;
(4) 为器械提供服务的人员;
(5) 提供的服务;和
(6) 任何测试和检测数据。
(c) 唯一器械标识 UDI。除了 ISO 13485 中第 7.5.1、7.5.8 和 7.5.9 条的要求外,还必须记录每个医疗器械或每批医疗器械的 UDI。
(d) 保密。制造商认为机密的记录可能会被标记,以帮助 FDA 确定是否可以根据本章第 20 部分的公共信息法规披露信息。
§ 820.40 [预留的]
§ 820.45 设备标签和包装控制
除了 ISO 13485 第 7.5.1 条的要求(通过引用并入,参见 § 820.7),生产和服务提供的控制,每个制造商必须建立和维护提供活动详细描述的程序,在器械的加工、储存、搬运、分销和适当情况下使用的常规条件下,以确保标签和包装的完整,检查,储存和操作。
(a) 制造商必须确保标签和包装在放行或储存之前已检查其准确性,如适用,包括以下内容:
(1) 正确的唯一器械标识 (UDI) 或通用产品代码 (UPC),或任何其他器械标识
(2) 有效期;
(3) 存储说明;
(4) 处理说明;和
(5) 任何额外的处理指引。
(b) 使用标签的发放必须按照 ISO 13485 的第 4.2.5 条记录。
(c) 制造商必须确保已建立并维护标签和包装操作以防止错误,包括但不限于在使用前及时检查标签和包装,以确保所有器械都有器械文档中要求的正确的标签和包装。此类标签检查的结果必须按照 ISO 13485 的第 4.2.5 条记录。

来源:Internet


