您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2024-10-16 08:30
2024.10.11, NMPA公布了《国家药监局关于发布境内生产药品再注册申报程序和申报资料要求的通告(2024年第38号)》。本次为正式稿,与2024.05.13发布的征求意见稿没有太大的区别。下面就笔者关注到的几个点,做一个梳理。
1、适用范围
1)境内生产药品上市许可持有人:含境内化学药品持有人、生物制品持有人和中药持有人;
2)境内化学原料药登记人:一般即为境内原料药生产企业。
2、再注册申请提交时间的考虑
根据《再注册申报程序》:
1.应当在药品批准证明文件(包括药品注册证书、化学原料药批准通知书、药品再注册批准通知书等)有效期届满前12个月至6个月期间提交再注册申请。
2.药品再注册审查审批时限为120日。
3.药品再注册批准通知书有效期自原药品批准证明文件有效期届满次日起算;原药品批准证明文件有效期届满后批准再注册的,药品再注册批准通知书有效期自省级药品监督管理部门批准再注册之日起算。
另外,NMPA曾于2024.05.13发布过《国家药监局综合司公开征求《关于发布境内生产药品再注册申报程序和申报资料要求的通告》意见》,要求药品批准证明文件有效期届满后不得继续生产:
“申请人在药品批准证明文件有效期届满前六个月内提出药品再注册申请的,或者存在中止受理、审查审批等情形的,药品批准证明文件有效期届满后不得继续生产;省级局准予再注册的,从批准再注册之日起,方可以生产”。
综合以上信息,再注册申请必须在有效期届满前6个月(即至少涵盖再注册审查审批时限:120日)提出,同时考虑到可能的补充资料等引起审评中止的情形,以防止出现批件效期的断档,导致不得生产(按假药论处)。
3、再注册申报方式
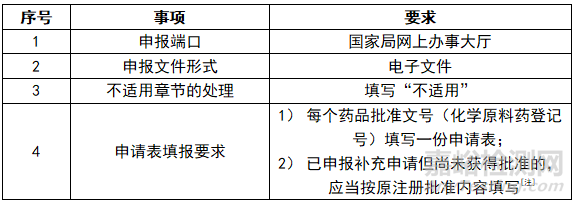
[注]再注册申请不得合并补充申请/备案事项,应独立平行申报。但在再注册申报资料“五、药品批准证明文件及其附件载明信息变化情况”中,仍然要求:“已提交补充申请但尚在审评审批阶段以及已完成备案但尚在审查阶段的变更情况也应当填报”。
4、证明性文件的提交
本次《境内生产药品再注册申报资料要求》对于证明性文件的提交要求,也明确“进行网络和内部核查核验的无需提供”。我们看到2020年7月1日CDE发布的《国家药监局药审中心关于发布《M4模块一行政文件和药品信息》的通告(2020年第6号)》中的证明性文件提交要求也几乎都是“内部核查”。
理论上,这些证明性文件都不需要提交了,但大部分注册同行们出于习惯、以及保险起见,一般还是会提供。
5、按照药品批准证明文件和药品监督管理部门要求开展相关工作的情况
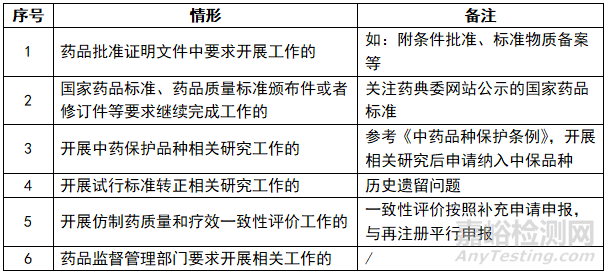
6、再注册期间涉及的证书类型
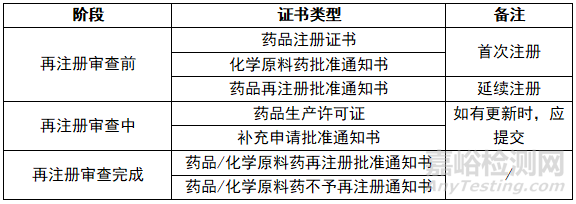
7、“药品再注册周期”内未生产品种的要求
1.申请人在药品再注册申请时主动说明药品生产状态;
2.申请人加强未开展商业化规模生产药品的管理,持续考察药品质量、疗效和不良反应情况,在恢复生产前对照现行的技术指导原则进行评估和研究,进行补充申请/备案/报告;
3.恢复生产时向省级药品监督管理部门提出现场检查、检验申请,现场检查、抽样检验合格的方可上市销售;
4.对于风险较高的注射剂,申请人还应当将后续两批样品送药品检验机构检验,样品检验合格的方可上市销售。

来源:注册圈


