相信负责原辅包登记申报的小伙伴们,平时遇到最多的不是新产品的首次登记,而是现有产品的登记变更。导致登记变更的情况很多,尤其是登记产品种类比较多的公司,比如随着稳定性数据积累到一定程度,有效期变更的工作便可进行;由于生产规模的扩大,需要增加车间或者转移新厂房,相应的变更也需要提上日程;由于五年一次的药典升版,相应的药典标准若有变化,则对应的辅料质量标准也许随之变更,于是变更质量标准的登记工作也得进行……变更的种类五花八门,变更的难易程度也是参差不齐,有的很简单,有的却要大费周折。更为尴尬的是,目前针对原辅包登记变更并没有官方的指导原则,以致很多时候都是参考一些相近的变更指导原则,再结合各自的知识和经验,评估确定变更的级别以及可能需要做得工作。由此可知,原辅包登记变革工作频繁而又比较依赖于经验的积累。
在这篇文章中,我将结合自己的工作经验,分享一个药用辅料登记变更的案例,变更内容包括有效期延长、质量标准升级、基本信息变化,同时补充全套登记资料,希望能给从事药用辅料登记注册的小伙伴们一些帮助。
一、案例基本情况概要
某药用辅料,已有在国外上市药品中使用的历史,且被中国药典、日本药典收录,因此可归属于2.4类中国药典已收载的辅料。
该药用辅料来源于植物,而非动物源或人源;拟登记给药途径为口服给药,而非注射剂、眼用制剂、吸入制剂,因此可归属于低风险药用辅料。
本品为早期通过技术审评,在没有颁发批准证明文件的情况下自动转A的品种。虽然实际生产过程中发生了不少变化,但多年来始终没人进行信息维护。
由于为期4年的稳定性试验已经完成,所以计划延长有效期至3年进行销售,于是便有了这次变更的任务。
二、情况梳理
经过梳理和沟通,进一步了解了目前的情况以及需要进行的工作,梳理如下:
1)4年的稳定性研究已完成,数据齐全,提交稳定性数据后,并以此进行有效期延长,数据链完整,资料齐全,风险不大。
2)本品2019年自动转A,但未按要求提交基于56号文附件一要求的全套登记资料,因此登记号下是空无资料的,导致后续的变更没有根基,需要先将全套资料补齐。如何补齐资料,是否可以和变更同步进行,是个需要考虑和评估的问题。
3)2019年至今,中国药典由2015版升级未2020版,导致之前批准的质量标准不再满足现今的要求,需要进行质量标准的升版。
4)由于市政原因,公司所在地址信息发生了变化,相应的营业执照中的地址信息与现有登记信息中的地址信息不一致,需要通过基础信息变更,使两者保持一致。
5)由于产能扩大,新增一个车间进行生产,希望能将新车间增加到登记资料中。
6)由于销售需要,新增了一些规格,希望能将新规格增加到登记资料中。
三、风险分析及策略拟定
经过上述信息梳理,我认为所有变更的前提是先补齐符合56号文附件一要求的全套登记资料,而变更的具体内容包括:
1)基于稳定性数据,延长有效期;
2)基于中国药典2020版,变更质量标准;
3)基于最新的资质资料,变更基础信息;
4)变更生产产地,增加车间;
5)变更规格,增加新规格。
于是开始思考以下问题:
1)补齐全套登记资料是否可以和变更合并进行?
2)这么多的变更是否可以分次进行,以免变更太多太大,影响登记状态?
3)这是变更如何分级,具体需要做哪些工作?
4)需要参考哪些法规或指导原则?
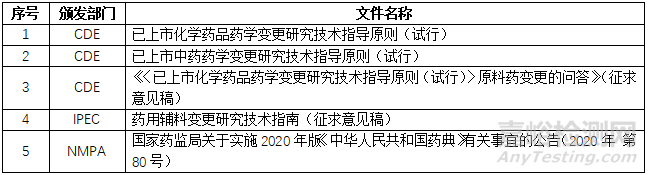
为此,我首先找来了以下相关的指导原则:
同时在CDE网站上提交了一个一般性技术问题咨询,最终拟定策略如下:
1)基于咨询结果,可以将变更与补交登记资料合并提供,预计决定这两个合并推进。
2)变更太多,为了不影响A状态,因此决定分次进行变更。优先进行有效期延长和质量标准提升的变更,顺便带上基础信息变更,原因在于前者是最紧急任务,后者是补交登记资料中需要的内容。针对稳定性数据未按照中国药典2020版要求检验的情况,取留样采用按注册标准和中国药典2020版标准分别检验进行对比说明。其次再考虑生产场地变更和规格变更,原因在于需补充的研究内容较多,耗时较长。
针对首先要进行的 “有效期延长+质量标准升级+基础信息修订”的变更登记,判定其变更分级为中级,原因如下:
1)首先,没有针对药用辅料自身变更分级的指导原则,相当于没有硬性要求,可以基于对产品本身,对由其制备而成的药品制剂,对最终服用药品制剂的患者的风险进行考虑分级。
2)有效期延长是基于切实的稳定性数据,科学合理而且信息充实。
3)质量标准基于中国药典升版属于官方要求的变更,而且中国药典作为法定标准,具有其权威性和普适性,因此只要产品满足其要求,质量风险可控。
4)申请人基本信息变更,属于非技术性变更,不会对产品质量带来风险。
具体需要进行的工作内容如下:
1)写一份变更说明,将上述变更的内容予以概况说明,便于审核老师快速了解整体情况;
2)按56号文附件一的要求,撰写全套登记资料;
3)收集稳定性数据及图谱,作为登记资料稳定性部分的内容;
4)对比原批准的注册标准及中国药典2020版,并基于以下要求拟定新的质量标准;
5) 选择留样,采用原注册标准和新拟定的标准进行对比检验,作为产品不仅能够满足新拟定质量标准,而且长期放置(PS:留样的放置条件与长期稳定性试验条件类似,但放置时间更长。)仍能满足新拟定的质量标准,从而进一步支持有效期延长。
四、执行过程
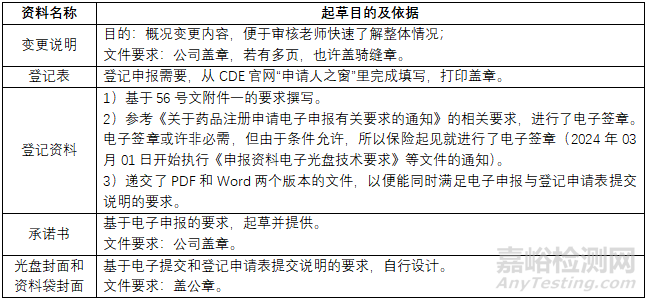
策略清晰,工作内容明确,执行起来就比较顺畅,整个过程完成的文件资料汇总如下:
五、经验总结
关于此次登记登记的经验总结如下:
1)变更的情况繁多、程度不同,信息评估和策略拟定至关重要,而其重点不仅仅在于对药用辅料本身的影响,还包括对制剂以及患者的影响。一些看着对辅料本身影响挺大的变更,可能对制剂或患者而言影响不大,因此可不用将其作为重大变更对待。
2)由于历史原因,未提交符合现行要求的登记资料的企业,可将登记变更与补交登记资料一并进行,可以不用分次进行。
3)变更内容很多时,可以不用一次性全部变更,可根据各自的情况分次变更。
4)宁多不缺,对于拿不准的地方,宁可多提供些,不要少提供,比如在对文件格式要求不清楚的时候,同时提交了PDF和Word版本的资料。