本篇聊聊医疗器械变更的基本流程,希望与大家产生共鸣。变更亦是持续改进的保证,可能发生在医疗器械的全生命周期。上市前的变更,对医疗器械性能和质量影响较小,涉及的范围也不大,比较容易控制。则应该对新增型号差异部分重点研究,必要时委托第三方机构进行检测。常见的有:新增生产设备、新增工艺装备、生产设备位置的变更、工艺参数的变更等。根据供应商的资质水平,可以适时增加或删减进货检验项目。伴随着产品或工艺的变更,相应的文件一般也会随着升版。企业负责人、管理者代表、质量负责人在当地的药监局都有备案。对供应商的管理,企业需要制定《供应商管理规定》类似的程序文件。还存在多非技术类的变更,如:公司名称的变更、公司住所的变更、生产地址的变更、生产环境的变更。这些需要企业另行制定,并且按规定向国家局或者省局备案登记。识别上述变更后,可以执行如下流程(可选择性或不限):相关部门发起变更申请,填写“变更申请表”,说明变更理由和变更的内容。相关部门对变更内容进行审评,由管理者代表批准后,方可执行变更措施。申请批准后,技术部、生产部或质检部等,执行变更措施。根据法律法规、公司内部文件、客户要求,及时通知相关方。必要时,还应针对变更内容,重点关注和监测:客户反馈信息以及不良事件。
确保变更后的医疗器械产品安全有效。
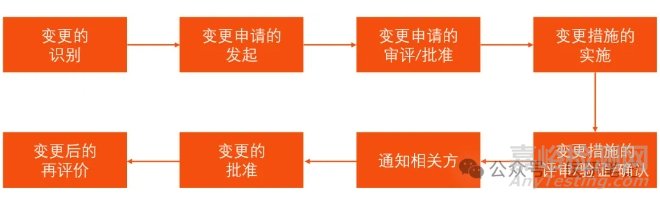
企业应当制定《变更管理控制程序》,或类似的管理文件。在该管理文件中,企业可以基于风险的管理的原则,结合自家的实际情况,将变更实行分类管理。比如一级变更(轻微)、二级变更(一般)、三级变更(重大)……
然后,针对不同级别的变更,分别采取不同的管理方式。



