您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2025-02-13 08:48
摘要
遗传毒性试验是药物非临床安全性评价的重要组成部分,主要用于药物早期致癌性风险评价。近年来,新靶点和创新药效结构的药物大量涌现,遗传毒性或致癌性结果呈阳性的创新药物数量大幅增加。然而,有研究显示细菌回复突变试验(Ames)结果为阳性的化合物中约1/3为非致癌物。本文回顾了致突变性与致癌性风险间的关联,Ames试验阳性结果的原因及当前国际上对Ames试验阳性结果的药物的监管要求,总结了可供选择的后续体内试验方法,并提出当药物在非临床安全性评价中Ames试验结果为阳性时的评价思路和机制研究策略,为药物研发及评价提供借鉴。
【关键词】 Ames试验;阳性结果;后续试验;转基因啮齿动物;Pig-a基因突变试验;下一代测序
遗传物质损伤与肿瘤的形成存在一定关联。近年来,新靶点和创新药效结构的药物大量涌现,遗传毒性或致癌性结果呈阳性的创新药物数量大幅增加。回顾性文献认为,遗传物质致断裂性风险通常与受试物的直接毒性有关[1],染色体或DNA的断裂性与剂量存在一定关联,可通过降低暴露量而避免染色体损伤风险。然而,致突变性作用则无剂量效应相关性,不可修复的突变形成后,可在不断复制过程中逐渐发展为肿瘤。在美国 《医师案头参考(1999)》 清单列出的 352 个非抗癌药物中,29%至少有一项遗传毒性试验为阳性,201种药品的标签中注明了遗传毒性和致癌性结果为阳性,38%的致癌性数据为阳性,8.3%的细菌回复突变试验(Ames 试验)结果为阳性[2]。又如,蒽醌类[3]和黄酮类[4]等中药中的常见成分Ames试验结果也为阳性,该类成分在我国已有上千年的服用历史。由此引发了以下两个问题:①当前在监管领域常用的遗传毒性试验方法是否可以科学地对创新药物的人体致癌性风险进行预测?②Ames试验结果为阳性的创新药物应采取怎样的评价策略?
本文聚焦细菌致突变性数据与致癌性风险的关联,综述了Ames试验阳性结果的原因,并提出了Ames试验阳性结果的后续研究策略,为药物遗传毒性研究和监管提供参考。
1、致突变性与致癌性
基因突变可增加人类患癌和遗传性疾病的风险,对健康产生深远影响。Hermann Muller最早于1927年报道了暴露于X射线的果蝇会产生新的遗传性状,证明了暴露于环境因素会改变生物体的基因组成[5]。自1953年DNA结构的发表及随后DNA聚合酶的描述,逐渐将环境暴露与突变和遗传变化联系起来[6]。20世纪70年代中期,通过啮齿动物致癌性研究数据表明大多数已知诱变剂均具有致癌性。
致癌物按照作用机制可分为对DNA造成直接损伤的遗传毒性致癌物和不直接造成DNA损伤的非遗传毒性致癌物,即表观遗传性致癌物。其中遗传毒性致癌物的致突变性作用,理论上没有阈值[7]。50多年来,致癌性与致突变性之间的密切关联一直是遗传毒性研究的驱动力。突变导致癌症重要的第一步是DNA损伤,致癌物可以导致DNA加合物的形成,诱发DNA的其他修饰,也会导致 DNA 链交联、断裂和染色体的缺失重排[8]。非遗传毒性致癌物无致突变性,但有阈值反应,在一定范围内不会产生不良影响,主要以DNA甲基化或组蛋白乙酰化两种方式影响某些基因的表达。本文聚焦致突变性和致癌性的风险评估。
2、Ames试验
Ames 试验由 Bruce N. Ames 于 1973 年首次报道,当前已成为全球范围内用于化学品和药物致突变性风险初步筛选的首选试验方法。该方法基于营养缺陷型鼠伤寒沙门氏菌(如TA97a、TA97、TA98、TA100、TA102、TA1535 和 TA1537 等)和大肠杆菌(WP2和WP2urvA)开展。在诱变剂作用下,缺陷基因反向突变,使细菌自主合成必需氨基酸并在培养基中形成可见菌落,从而通过菌落计数来评价受试物的致突变性风险。鉴于Ames试验简单灵活、成本效益好,并建立了经验证的大型数据库,当前已基于化合物的结构特征、Ames 试验数据和计算拓展分析技术开发了多种体外致突变性风险检测模型。如Hansen 等[9]构建的包含6500种化学物质以及沙门氏菌检测数据和结构信息的基准数据集,以及Vian等[10]开发的可评估S9组分代谢活化对Ames计算机模型的影响的预测模型等。
2.1 Ames试验阳性药物的监管现状
通常Ames试验结果呈阳性的化合物被视为存在体内致突变性风险或致癌性风险。美国食品药品监督管理局(Food andDrug Administration,FDA)允许Ames试验阳性药物在健康受试者中进行单次给药试验。某些监管机构则允许超过单次的使用,如英国药品和健康产品管理局(Medicines and HealthcareProducts Regulatory Agency,MHRA)认为在健康受试者中使用存在遗传毒性的受试物时需要提供具体的理由,当理由充分(如基于毒理学关注阈值的风险评估数据、药物半衰期数据、人临床拟用剂量范围等)时,可在临床给予单剂量的 Ames 阳性药物。如无充分的理由,单剂量给予周期不可超过1周。此外,人用药品注册技术要求国际协调会(International Council forHarmonization of Technical Requirement for Pharmaceuticalsfor Human Use,ICH)发布的 ICH M3(R2)指导原则[11]指出如药物无警示结构,可以100 μg/d的剂量分5次给予,该剂量为临床试验中使用药理活性剂量的1/1 000~1/10以下。德国联邦药品 和 医 疗 器 械 机 构 (Bundesinstitut für Arzneimittel undMedizinprodukte,BfArM)也允许在健康受试者中使用微剂量的Ames 阳性药物。然而,加拿大卫生部则认为如果没有后续研究证明该类药物在体内无致突变性,则不允许其在健康受试者中进行临床试验。日本监管部门也不允许在健康受试者中使用Ames阳性药物。中国自加入ICH以来,对Ames阳性药物的监管主要参考ICH指导原则。
2.2 Ames试验阳性药物的机制探索
Ames试验的阳性结果与致癌性风险密切相关,但Ames试验结果也可能是由多种因素影响产生的假阳性结果,因此对Ames阳性结果的机制探索十分重要。
2.2.1 杂质
Ames试验阳性结果可能由低浓度的致突变性杂质引起,这些杂质在ICH M7(R2)指导原则中属于1类和2类杂质。对于已知具有致突变性和致癌性的1类杂质,首先考虑去除,如果无法去除,则应建立基于未观察到作用水平(noobserved effect level,NOEL)致癌性数据的限度数值。监管评估的常规方式是根据致癌性剂量反应数据采用线性低剂量外推法,即以诱导50%肿瘤发生率的剂量(TD50)的五万分之一为摄入量,将肿瘤的发生风险控制在十万分之一。对于已知致突变性但致癌性未知的2类杂质,如存在阈值相关机制,以每日允许暴露量(permitted daily exposure,PDE)求限度值;如无阈值相关机制的证据,根据毒理学关注阈值(threshold of toxicologicalconcern,TTC)确立限度。如2007年在抗HIV药物Viracept中发现了遗传毒性杂质EMS,Gocke 等[12]开展多剂量的转基因小鼠致突变性试验,并结合微核试验和组织中加合物检测,得到25 mg/(kg·d)为EMS致突变效应的起始阈值,且该剂量下重复给药无蓄积效应。Viracept 中 EMS 的人体最大暴露剂量为0.055 mg/(kg·d),远低于致突变作用阈值。故认为使用Viracept的患者不会因接触 EMS而出现任何额外的毒理不良反应。
但有些高致突变致癌物被称为“关注队列”,即使摄入量低于TTC,理论上仍会具有高致癌风险,主要包括黄曲霉毒素样化合物、N-亚硝基化合物和烷基氮氧化合物。其中N-烷基亚 硝 胺 例 如 N-亚 硝 基 二 甲 胺 (NDMA) 和 N-亚 硝 基 二 乙 胺(NDEA),是已知的对大鼠具有遗传毒性的致癌物质。这类物质具有非线性剂量反应和实际阈值,风险评估不能使用TTC。遵循ICH建议,Johnson等[13]使用已发表的啮齿动物癌症生物测定和体内致突变性数据计算NDMA对癌变和突变的PDE分别为每人 6.2 和 0.6 μg/d,NDEA 对癌变和突变的 PDE 分别为每人 2.2和0.04 μg/d,均高于简单线性外推得出的可接受的每日摄入量(NDMA为96 ng,NDEA为26.5 ng)。这为使用基准方法的PDE计算提供了更可靠的暴露限值评估并且可以更好地告知患者的风险。
2.2.2 化学结构
药物的结构成分之间相互干扰可能产生毒性,从而使 Ames 结果为阳性。“警示结构”一词由 Ashby[14]在1985年定义致癌性的结构基础时引入,它是与化学物质的致癌和致突变特性相关的分子结构或活性基团,不仅对潜在致癌物的分类非常有帮助,而且对理解遗传毒性机制也很重要。Perez-Garrido 等[15]提出了一系列针对致突变性的警示结构,表明致突变性与疏水性和分子体积之间存在相关性,创建的模型一致性为86%,正确分类了95%的致突变物质。Amberg等[16]证明在使用DMSO 作为媒介的Ames 试验中,产生阳性结果的18种酰基或磺酰卤化学品中有15种在使用水为媒介时产生阴性结果。这可能是由于酰基或磺酰卤在DMSO中进行酰化而产生卤代甲基硫醚,也可能是在水中分别水解为非诱变碳酸和卤化氢或磺酸和卤化氢。由此可以得出酰基或磺酰卤是Ames致突变性的警示结构。
国际上提出采用警示结构作为区分普通杂质和遗传毒性杂质的重要依据,这一概念也成为遗传毒性警示结构以及定量构效关系(quantitative structure activity relationship,QSAR)模型的基础。ICH M7 指南允许在试验数据不足时使用计算机 QSAR方法来预测细菌致突变性,使用两种互补的方法即基于专家规则和基于统计搜索是否存在警示结构是将杂质分类为3、4或5类的第一步。在基于规则的系统中,定性预测基于化学品的警示结构,因为警示结构周围的电子密度和空间环境及分子的大小和形状等可能影响预测结果。在输入药物特有的信息后,QSAR模型会自动与已有数据比较,根据结构相似程度,判断是否存在警示结构。将警示结构与QSAR模型相结合可以提高毒性预测的准确性,减少未来候选药物产生不良毒性作用的可能性[17]。
2.2.3 代谢产物
细菌和哺乳动物具有不同的新陈代谢能力,与哺乳动物细胞相比,细菌能够有效地将硝基和偶氮化合物活化为亲电代谢物。为了提高其代谢能力,在Ames试验体系中常加入哺乳动物肝 S9,但在有利于高水平氧化酶代谢的条件下,Ames试验结果可能为阳性。Ames试验阳性药物相关代谢物与2年啮齿类动物致癌性生物测定具有高度相关性,如Ames试验阳性药物相关代谢物低于全身总暴露量的10%,则应考虑原料药剂量、代谢物中活性官能团性质等进行后续测试。根据ICH M3 (R2)指南,如果Ames试验阳性药物相关代谢物大于全身 总 暴 露 量 的 10% , 则 可 能 以 处 理 药 物 活 性 成 分 (activepharmaceutical ingredients,API)类似的方式处理。药物代谢物在结构上存在致突变性,则需要特别关注。如酰胺形成的键对酶或酸催化的水解具有潜在的不稳定性,从而产生游离的芳香胺,后者可作为杂质或代谢物或降解物出现。在这种情况下,既将其作为杂质,又要将其作为代谢物进行Ames试验。
2.2.4 氧化剂
多种化学物质可通过产生活性氧(reactiveoxygen species,ROS)诱导 DNA 损伤,如农药、溶剂(如氯仿、四氯化碳和酚类)、儿茶酚和儿茶酚胺、雌激素、金属和苯并[a]芘等。当产生的ROS浓度超过抗氧化剂清除能力时,对细胞有害并导致DNA、蛋白质和脂质等细胞大分子受损。最终,ROS的积累会引起氧化应激,导致细胞死亡[18]。一些药物在Ames试验中呈阳性但在哺乳动物细胞试验中结果为阴性,此时需考虑是否与氧化应激作用机制有关。Ames 菌株应对特定氧化应激的能力各不相同,TA97、TA100、TA102 对氧化应激更敏感。Kirkland等[19]在不影响S9的代谢活化条件下,在Ames试验系统中添加抗氧化剂。氧化剂乙二醛在无代谢活化条件下导致TA100 和 TA1535 菌株的回复菌落数大幅增加,结果均为阳性,提示其有致突变性。经超氧化物歧化酶预处理后,乙二醛诱导的TA100回复突变菌落数减少66%,TA1535减少40%。经过氧化氢酶预处理后,TA100减少60%,TA1535减少21%。
3、体内致突变性试验
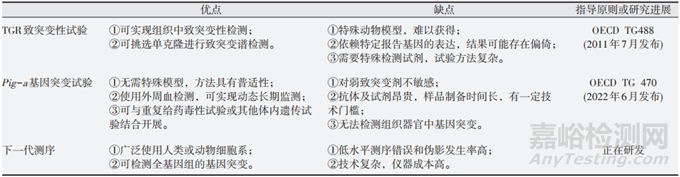
体内遗传毒性试验通常是标准试验组合的一部分,可以提供影响化合物遗传毒性的其他相关因素如吸收、分布、代谢和排泄等。在 Ames 试验阳性结果的后续体内研究试验选择中,Zeller等[20]建议首先开展骨髓和血液微核试验,并结合涵盖其他相关组织的彗星试验。Kirkland 等[21]发表的一项关于体内遗传毒性试验对人类致癌物性能评估的研究表明,骨髓微核和彗星试验相结合,可以检出约95%的人类致癌物。虽然彗星和微核试验对可能最终导致基因突变的DNA损伤都很敏感,但这两种检测方法的检测对象为大范围的遗传物质损伤,不能直接检测基因突变。ICH S2(R1)指出,对于 Ames 试验阳性药物,后续试验应以评估致突变性终点为基础,其目的是确定体外基因突变结果是否与体内基因突变结果相关,即与体内结果具有生物学相关性。OECD于2011年颁布的TG488指导原则提供了使用转基因啮齿动物模型评估生殖细胞和体细胞致突变性的建议,2020 年发布相关指导原则,指出当 Ames 试验结果不明确时,体内Pig-a基因突变试验可作为后续试验方法选择,以下分别对这两种方法进行阐述。
3.1 转基因啮齿动物致突变性试验
转基因啮齿动物(transgenic rodent,TGR)致突变性试验作为经典的体内遗传毒性评价方法至今已有20多年的历史。该方法可利用肝、肾等不同组织对基因突变进行检测,突破了体内遗传毒性评价方法仅使用造血组织为检测终点的局限。TGR主要利用 MutaTM小鼠、Big Blue®小鼠和大鼠、LacZ 质粒小鼠和 gptdelta 小鼠和大鼠,检测由复制过程中碱基配对或结合错误或DNA序列重排引起的小规模遗传损伤。在啮齿动物给药中最高剂量应为最大耐受剂量(maximum tolerated dose,MTD),可以根据死亡率、体质量减轻等标准终点来确定或通过临床观察选择。Luan 等[22]建立了一种对肝脏细胞色素 P450 还原酶无效的gpt delta 转基因小鼠模型,以评估4-甲基亚硝基胺基-1-3-吡啶 基-1-丁 酮 [4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone,NNK]诱发的基因突变。试验显示NNK不能在没有P450代谢激活的情况下诱导肝脏突变,但可以通过肝脏P450独立的机制诱导肺部突变,模型有助于确定肝脏与肝外 P450 介导的突变作用,以及P450与其他生物转化酶在遗传毒性致癌物激活中的作用。TGR 原则上可以与其他体内遗传毒性试验相结合,与Pig-a基因突变试验相结合,有助于减少费用和啮齿动物的使用,是检测Ames阳性结果后续工作的潜在方法。但转基因动物成本较高,某些特定的动物品系依赖从国外进口,不仅价格昂贵,而且进口周期漫长且渠道单一,不具有普适性。此外,TGR检测依赖特定报告基因的表达,检测结果可能存在偏倚。试验需要特殊检测试剂,价格昂贵,技术门槛高。转基因动物的检测结果外推至人体还存在一定差距。
3.2 体内Pig-a基因突变试验
体内Pig-a基因突变试验是考察外源物质导致的体内致突变风险的最有前途的试验方法。该方法主要通过流式细胞仪对细胞进行免疫荧光标记和分析,随时监测糖基磷脂酰肌醇(glycosyl phosphatidyl inositol,GPI)锚相关表面蛋白的表达缺失情况,作为Pig-a基因突变的指标。Pig-a试验以外周血作为检测对象,突变细胞在外周血中出现的时间取决于细胞和GPI锚的周转率,以及特定细胞类型从骨髓到外周的运输时间。Nishimura 等[23]报告了在阵发性睡眠性血红蛋白尿(paroxysmalnocturnal hemoglobinuria,PNH)患者的Pig-a基因中发现的一系列体细胞突变,包括碱基置换以及不同大小的插入和缺失,几乎所有病例的PNH诊断都与Pig-a基因突变有关。Dertinger等[24]进一步将高通量免疫磁性筛选技术引入 Pig-a 基因突变试验,从而有效提高了细胞分析的数量与统计功效。OECD在2022年发布了体内Pig-a基因突变试验指导原则TG470,给体内基因突变的实验设计带来了更多选择。近年来,以TK6、MCL-5以及L5178Y等哺乳动物细胞系开展的Pig-a基因突变试验也取得了一些进展[25],但目前尚处于研发阶段。与TGR基因突变检测不同,Pig-a检测不局限于特定的动物品系,可以与重复剂量毒理学研究和其他遗传毒理学试验相结合,符合3R原则,同时只需采取微量外周血样本,不会影响其他终点的评估。但Pig-a也具有一定局限性,它只能在红细胞和网织红细胞中进行。因此,需证明目标组织对药物或其代谢物的相关暴露。IWGT工作组建议可以将Pig-a与下一代测序或深度测序相结合,从而形成可应用于大鼠以外试验体系和其他造血细胞的新方法[26]。
4、下一代测序技术
在使用具有潜在遗传毒性的药物治疗后,细胞基因突变频率对于危害评估十分关键。已经建立的体外体内遗传毒性试验和转基因动物模型不能提供整个基因组序列完整性信息。下一代测序(next generation sequencing,NGS)技术提供了多种研究基因组的方法,包括整个基因组、外显子组和转录子组的测序。NGS技术不依赖任何特定的基因或细胞系,具有高通量和大规模并行测序的能力,可以同时筛选多个样本中的各种基因组变化。尽管NSG明显优于传统Sanger测序,但在DNA制备、扩增和测序过程中经常出现低水平测序错误和伪影,可导致高达1%的人工突变频率,远远高于正常组织的正常每个核苷酸突变频率(<1×10-7),且在某些序列背景下,可能会更高。因此,为了检测更低频率的突变并提高测序准确度,需要开发更灵敏的NGS技术。
目前认为降低低频测序错误率最有效的策略是分子一致性测序,基于单链或互补链对同一模板来源的多个拷贝进行错误校正。Schmitt 等[27]通过对 DNA 互补链添加分子标记,开发了Duplex Sequencing方法,测序错率低至1×10-7,但测序深度和成本高,所需拷贝数多,不适用于大型基因组。Hoang等[28]在Duplex Sequencing基础上开发BotSeq,对PCR扩增后的文库进行稀释达到提高测序效率的目的,可用于全基因组测序。Abascal等[29]又在BotSeq基础上开发了NanoSeq,引入特定限制性内切酶和ddBTPs,降低文库制备损伤和末端修复错误,测序效率高,错误率降低至 1×10-8。为了避免 PCR 扩增引入的伪影,2020年,You等[30]开发了一种无PCR的缩短双链独立一致性的测序方法 PECC-Seq(paired-end complementary consensussequencing),以剪切点为内源标记,进一步简化文库制备及后续生物信息学分析,理论错误率可降低至1×10-9。在全基因组范围内的低频测序表现出较高的准确度和较好的成本效益,在临床液体活检、癌症研究等领域具有潜在的应用前景。应进一步对新的低频测序方法进行研究,以继续扩大测序应用范围。
5、小 结
化合物的Ames试验结果为阳性时,首先应分析阳性结果产生的原因。鉴于遗传毒性试验方法基于特定检测终点而形成的特点,应充分考虑Ames试验结果是否与细菌试验体系的特殊性或受试物的特殊作用机制有关,是否与细菌特定的代谢活化体系有关,是否与存在特定的警示结构有关。建议使用相关结构化合物或在调整试验条件后开展比较研究,以判断是否可排除其人体致癌性风险。后续可选择以基因突变为检测终点的评价方法包括,小鼠淋巴瘤细胞试验、体内Pig-a基因突变试验和TGR 致突变性试验等(表1)。当前下一代测序技术国际上正在研发阶段。除上述以致突变性为检测终点的试验方法外,也可考虑开展转基因小鼠6个月致癌性试验,从而在有限的时间和经济成本内获得TD50数据用于风险评估。此外,也可开展多剂量的体内致突变性/致癌性试验,验证其是否具有作用阈值。开展体内研究时可伴随开展药代/毒代动力学、组织加合物检测、突变谱检测等机制研究,从而进一步了解其致突变性作用特点,为同类药物的设计与合成提供重要参考。
▲表1-致突变性检测方法比较
综上所述,遗传毒性试验是药物非临床安全性评价的重要组成部分,主要用于药物早期致癌性风险评价。本文回顾了致突变性与致癌性风险间的关联,以及Ames试验阳性结果的原因及当前国际上对Ames试验为阳性结果的药物监管要求,总结了可供选择的后续体内试验方法,并提出当药物在非临床安全性评价中Ames试验结果为阳性时的评价思路和机制研究策略,为药物研发及评价提供借鉴。
参考文献
[1]DISHOTSKY N I, LOUGHMAN W D, MOGAR R E, et al. LSD andgenetic damage[J]. Science, 1971, 172(3982): 431-440.
[2]SNYDER R D, GREEN J W. A review of the genotoxicity of marketed
pharmaceuticals[J]. Mutat Res, 2001, 488(2): 151-169.
[3]王亚楠, 王雪, 汪祺, 等. 基于毒理学软件和细菌回复突变试验的大黄素型蒽醌基因突变风险评价[J]. 药物评价研究, 2022, 45(7): 1240-1247.
[4] RESENDE F A, VILEGAS W, DOS SANTOS L C, et al. Mutagenicity of
flavonoids assayed by bacterial reverse mutation (Ames) test[J].Molecules, 2012, 17(5): 5255-5268.
[5]MULLER H J. Artificial transmutation of the gene[J]. Science, 1927, 66
(1699): 84-87.
[6]BESSMAN M J, LEHMAN I R, SIMMS E S, et al. Enzymatic synthesis
of deoxyribonucleic acid. II. General properties of the reaction[J]. J Biol
Chem, 1958, 233(1): 171-177.
[7] KIRSCH-VOLDERS M, AARDEMA M, ELHAJOUJI A. Concepts of threshold in mutagenesis and carcinogenesis[J]. Mutat Res, 2000, 464
(1): 3-11.
[8]PLOŠNIK A, VRAČKO M, DOLENC M S. Mutagenic and carcinogenic
structural alerts and their mechanisms of action[J]. Arh Hig Rada
Toksikol, 2016, 67(3): 169-182.
[9]HANSEN K, MIKA S, SCHROETER T, et al. Benchmark data set for in
silico prediction of Ames mutagenicity[J]. J Chem Inf Model, 2009, 49
(9): 2077-2081.
[10] VIAN M, RAITANO G, RONCAGLIONI A, et al. In silico model formutagenicity (Ames test), taking into account metabolism[J].Mutagenesis, 2019, 34(1): 41-48.
[11] ICH. Guidance on Nonclinical Safety Studies for the Conduct of HumanClinical Trials and Marketing Authorization for Pharmaceuticals [EB/OL]. (2017-02-15) https: //database.ich.org/sites/default/files/M3_R2_Guideline.pdf.
[12] GOCKE E, MÜLLER L. In vivo studies in the mouse to define a
threshold for the genotoxicity of EMS and ENU[J]. Mutat Res, 2009, 678
(2): 101-107.
[13] JOHNSON G E, DOBO K, GOLLAPUDI B, et al. Permitted daily exposure limits for noteworthy N-nitrosamines[J]. Environ Mol Mutagen,2021, 62(5): 293-305.
[14] ASHBY J. Fundamental structural alerts to potential carcinogenicity ornoncarcinogenicity[J]. Environ Mutagen, 1985, 7(6): 919-921.
[15] PÉREZ-GARRIDO A, HELGUERA A M, RODRÍGUEZ F G, et al.QSAR models to predict mutagenicity of acrylates, methacrylates and alpha, beta-unsaturated carbonyl compounds[J]. Dent Mater, 2010, 26(5): 397-415.
[16] AMBERG A, HARVEY J S, CZICH A, et al. Do carboxylic/sulfonicacid halides really present a mutagenic and carcinogenic risk asimpurities in final drug products?[J]. Org Process Res Dev, 2015, 19(11): 1495-1506.
[17] ALVES V, MURATOV E, CAPUZZI S, et al. Alarms about structural
alerts[J]. Green Chem, 2016, 18(16): 4348-4360.
[18] WANG E Y, HUANG Y, DU Q Y, et al. Silver nanoparticle induced
toxicity to human sperm by increasing ROS(reactive oxygen species)
production and DNA damage[J]. Environ Toxicol Pharmacol, 2017, 52:
193-199.
[19] KIRKLAND D, ZEIGER E, MADIA F, et al. Can in vitro mammalian
cell genotoxicity test results be used to complement positive results in the Ames test and help predict carcinogenic or in vivo genotoxic activity? I. Reports of individual databases presented at an EURL ECVAM Workshop[J]. Mutat Res Genet Toxicol Environ Mutagen, 2014, 775/776: 55-68.
[20] ZELLER A, BRIGO A, BRINK A, et al. Genotoxicity assessment of drug metabolites in the context of MIST and beyond[J]. Chem Res Toxicol, 2020, 33(1): 10-19.
[21] KIRKLAND D, LEVY D D, LEBARON M J, et al. A comparison of transgenic rodent mutation and in vivo comet assay responses for 91 chemicals[J]. Mutat Res Genet Toxicol Environ Mutagen, 2019, 839: 21-35.
[22] LUAN Y, XING G Z, QI X M, et al. The application of hepatic P450 reductase null gpt delta mice in studying the role of hepatic P450 ingenotoxic carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
induced mutagenesis[J]. Arch Toxicol, 2012, 86(11): 1753-1761.
[23] NISHIMURA J, MURAKAMI Y, KINOSHITA T. Paroxysmal nocturnal
hemoglobinuria: an acquired genetic disease[J]. Am J Hematol, 1999, 62
(3): 175-182.
[24] DERTINGER S D, PHONETHEPSWATH S, WELLER P, et al. Interlaboratory pig-a gene mutation assay trial: studies of 1, 3-propanesultone with immunomagnetic enrichment of mutant erythrocytes[J].Environ Mol Mutagen, 2011, 52(9): 748-755.
[25] 李若婉, 周长慧, 黄鹏程, 等. 基于TK6细胞的体外PIG-A基因突变检测方法的建立[J]. 癌变·畸变·突变, 2019, 31(3): 242-248.
[26] BEMIS J C, HEFLICH R H. In vitro mammalian cell mutation assays
based on the pig-a gene: a report of the 7th international workshop on
genotoxicity testing (IWGT) workgroup[J]. Mutat Res Genet Toxicol
Environ Mutagen, 2019, 847: 403028.
[27] SCHMITT M W, KENNEDY S R, SALK J J, et al. Detection of ultra-rare mutations by next-generation sequencing[J]. Proc Natl Acad Sci U S A, 2012, 109(36): 14508-14513.
[28] HOANG M L, KINDE I, TOMASETTI C, et al. Genome-wide quantification of rare somatic mutations in normal human tissues using massively parallel sequencing[J]. Proc Natl Acad Sci USA, 2016, 11(35): 9846-9851.
[29] ABASCAL F, HARVEY L M R, MITCHELL E, et al. Somatic mutation
landscapes at single-molecule resolution[J]. Nature, 2021, 593(7859):
405-410.
[30] YOU X Y, THIRUPPATHI S, LIU W Y, et al. Detection of genome-wide low-frequency mutations with Paired-End and Complementary Consensus Sequencing (PECC-Seq) revealed endrepair-derived artifacts as residual errors[J]. Arch Toxicol, 2020, 94(10): 3475-3485.

来源:中国知网


