您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2025-02-26 20:27
摘 要: 建立了固相萃取-液相色谱-串联质谱法测定毛发中45种毒品及代谢物。毛发样品经冷冻粉碎,研磨成粉末后,以1 mol/L硫酸锌-甲醇溶液(体积比为4∶6)超声提取,离心后,上清液分散至pH为6的纯水中,经HLB固相萃取柱分离富集,采用液相色谱质谱联用仪对45种毒品及其代谢物进行测定。以保留时间和特征离子对定性,以内标法定量分析。目标物的质量浓度在0.05~10 ng/mL范围内与色谱峰面积线性关系良好,相关系数均不小于0.999 0,检出限为0.067 1~233 pg/mg。目标物的平均回收率为60.2%~120%,测定结果的相对标准偏差为0.9%~12%(n=6)。该方法操作简单、灵敏度高、抗干扰能力强,满足毛发中毒品检测标准的要求,适用于毛发中45种毒品及代谢物的同时分析。
关键词: 固相萃取; 液相色谱-串联质谱法; 毛发; 毒品
在全球范围内,毒品都是一个严重而复杂的社会问题,对个人、家庭和社会造成了巨大危害,破坏了人类的健康、安全与社会稳定。因此,毒品检测作为应对这一问题的重要手段,其主要目标在于识别和监测个人是否使用了被禁止的药物。毒品检测技术广泛应用于体育竞技、就业背景调查、法律执法和临床诊断等多个领域[1]。目前常用的毒品检测技术包括对尿液、口腔、血液和毛发等样本的分析,以及现代生物传感器和化学分析技术[2-4]。毛发检测作为一种有效的毒品筛查方法,通过分析个体毛发中的代谢产物,能够长期追溯毒品滥用的历史。与尿液或血液检测相比,毛发检测具有更长的检测窗口,通常可覆盖数月至数年[5]。这一方法的原理在于毒品及其代谢物通过血液进入体内后,会沉积在毛囊中,因此毛发成为保存个体毒品暴露历史的生物标本。毛发检测不仅具有高度的准确性,而且相对非侵入性,为毒品滥用监测提供了一种可靠的手段。这使得毛发检测在毒品滥用监测领域日益受到重视。
毛发中毒品常用的分析方法主要包括气相色谱-质谱联用(GC-MS)法[6-7]、三重四级杆型气相色谱质谱联用(GC-MS/MS)法、液相色谱质谱联用(LC-MS)法[8-12]。其中,气相色谱质谱法分析时间长,且需要将目标化合物衍生后才能进样,操作繁琐。而液相色谱串联质谱法避免了衍生步骤,分析时间更短,灵敏度更高,在毒品分析中应用最为普遍[13-18]。
现有的司法鉴定检测标准如SF/Z JD0107025—2018《毛发中15种毒品及代谢物的液相色谱-串联质谱检验方法》,只采用甲醇超声提取的方式对毛发中的毒品进行提取,对于阳性样品,标准只要求复测,排查假阳性风险的手段较为单一。
笔者建立了一种固相萃取与液相色谱-串联质谱联用的方法测定毛发中45种毒品及代谢物。通过加入硫酸锌甲醇溶液去除毛发中蛋白的干扰。该方法灵敏度高、操作简单、抗干扰能力强,适用于实际案情里测定毛发中毒品及其代谢产物。
1. 实验部分
1.1 主要仪器与试剂
液相色谱质谱联用仪:LCMS-8045型,日本岛津公司。
超声清洗仪:JP-080s型,广东洁盟超声实业有限公司。
电子天平:AUW120D型,感量为0.000 1 g,日本岛津公司。
多样品组织研磨机:Tissuelyser-24型,上海净信实业发展有限公司。
12位固相萃取真空装置:上海安谱实验科技股份有限公司。
无水硫酸锌:分析纯,上海麦克林生化科技股份有限公司。
氢氧化钠:分析纯,天津大茂化学试剂厂。
盐酸:分析纯,广州化学试剂厂。
甲醇、乙腈:色谱纯,德国默克公司。
HLB固相萃取柱:规格分别为(1)150 mg(6 mL);(2)500 mg(6 mL),美国沃特世公司。
MCX固相萃取柱:150 mg(6 mL),美国沃特世公司。
实验用水为经Milli-Q净化系统过滤的超纯水。
毛发中36种化合物混合标准溶液:含有苄基哌嗪、吗啡、甲卡西酮、苯丙胺、甲基苯丙胺、乙卡西酮、3,4-亚甲二氧基甲卡西酮、可待因、3,4-亚甲基二氧基苯丙胺、3,4-亚甲二氧基甲基苯丙胺、对甲氧基甲基苯丙胺、O6-乙酰吗啡、去甲氯胺酮、氯胺酮、苯甲酰爱康宁、曲马多、海洛因、可卡因、亚甲基二氧吡咯戊酮、乙酰芬太尼、奥芬太尼、丙烯酰芬太尼、芬太尼、呋喃芬太尼、异丁酰芬太尼、4-氟丁酰芬太尼、戊酰基芬太尼、硝甲西泮、地西泮、四氢大麻酸、三唑仑、去甲乙酰芬太尼、艾司唑仑、氯硝西泮、右美沙芬、哌替啶各成分的质量浓度均为100 μg/mL,美国Cerillant公司。
毛发中9种化合物混合标准溶液:含有卡西酮、甲氧麻黄酮、利他林、2-亚乙基-1,5-二甲基-3,3-二苯基吡络烷、美沙酮、安眠酮、氟硝西泮、阿普唑仑、去甲芬太尼各成分的质量浓度均为1 000 μg/mL,美国Cerillant公司。
38种化合物混合内标溶液:含有苄基哌嗪-D7、吗啡-D3、卡西酮-D5、甲卡西酮-D3、苯丙胺-D5、甲基苯丙胺-D5、3,4-亚甲二氧基甲卡西酮-D3、可待因-D6、3,4-亚甲基二氧基苯丙胺-D5、3,4-亚甲二氧基甲基苯丙胺-D5、甲氧麻黄酮-D3、O6-乙酰吗啡-D3、去甲氯胺酮-D4、氯胺酮-D4、苯甲酰爱康宁-D3、利他林-D9、曲马多-D3、海洛因-D9、可卡因-D3、丙烯酰芬太尼-D5、芬太尼-D5、呋喃芬太尼-D5、异丁酰芬太尼-13C6、2-亚乙基-1,5-二甲基-3,3-二苯基吡络烷-D3、美沙酮-D3、戊酰基芬太尼-D5、安眠酮-D7、氟硝西泮-D7、地西泮-D5、阿普唑仑-D5、四氢大麻酸-D3、三唑仑-D4、去甲乙酰芬太尼-13C6、去甲芬太尼-D5、艾司唑仑-D5、氯硝西泮-D4、右美沙芬-D3、哌替啶-D4各成分的质量浓度均为100 μg/mL,美国Cerillant公司。
1.2 仪器工作条件
1.2.1 色谱仪
色谱柱:Agilent SB-AQ C18柱(150 mm×2.1 mm,1.8 μm,美国安捷伦科技有限公司);流动相:A相为0.1%(体积分数)甲酸溶液,B相为乙腈,流量为0.3 mL/min;柱温:35 ℃;进样体积:10 μL;洗脱方式:梯度洗脱,洗脱程序见表1。
表1 梯度洗脱程序
Tab. 1 Gradient elution program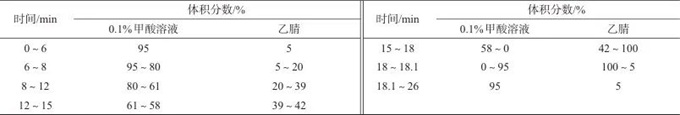
1.2.2 质谱仪
离子源:电喷雾离子源(ESI);扫描方式:正离子模式;检测方式:多反应监测(MRM);接口电压:4 kV,接口温度:300 ℃;脱溶剂温度:526 ℃;DL管温度:250 ℃;雾化气流量:3.0 L/min;加热气流量:10.0 L/min;加热块温度:400 ℃;干燥气流量:10.0 L/min。目标化合物及内标的质谱参数见表2。
表2 目标化合物及内标的质谱参数
Tab. 2 Mass spectrum parameters of the target compound
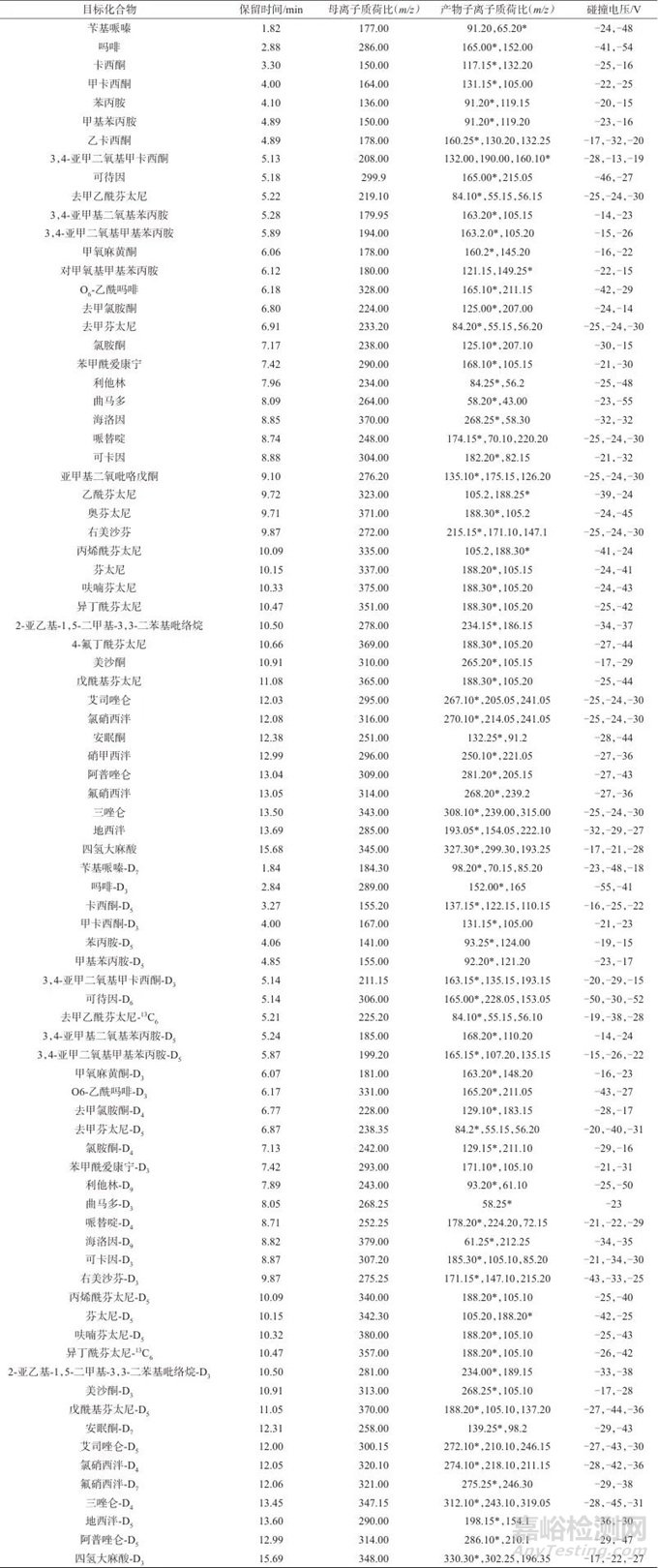
注:*为定量离子。
1.3 溶液配制
将45种待测化合物根据仪器响应分为以下3组:苄基哌嗪、吗啡、卡西酮、甲卡西酮、乙卡西酮、可待因、3,4-亚甲基二氧基苯丙胺、O6-乙酰吗啡、海洛因、硝甲西泮、地西泮、阿普唑仑、四氢大麻酸、三唑仑、艾司唑仑、氯硝西泮和哌替啶为A组;甲基苯丙胺、3,4-亚甲二氧基甲卡西酮、3,4-亚甲二氧基甲基苯丙胺、甲氧麻黄酮、对甲氧基甲基苯丙胺、去甲氯胺酮、氯胺酮、苯甲酰爱康宁、利他林、曲马多、可卡因、亚甲基二氧吡咯戊酮、乙酰芬太尼、奥芬太尼、丙烯酰芬太尼、芬太尼、呋喃芬太尼、异丁酰芬太尼、2-亚乙基-1,5-二甲基-3,3-二苯基吡络烷、4-氟丁酰芬太尼、美沙酮、戊酰基芬太尼、安眠酮、去甲乙酰芬太尼、去甲芬太尼和右美沙芬为B组;苯丙胺和氟硝西泮为C组。
将38种内标根据仪器响应分为以下两组:苄基哌嗪-D7、吗啡-D3、卡西酮-D5、甲卡西酮-D3、苯丙胺-D5、可待因-D6、3,4-亚甲基二氧基苯丙胺-D5、O6-乙酰吗啡-D3、海洛因-D9、氟硝西泮-D7、地西泮-D5、阿普唑仑-D5、四氢大麻酸-D3、三唑仑-D4、艾司唑仑-D5、氯硝西泮-D4和哌替啶-D4为D组;甲基苯丙胺-D5、3,4-亚甲二氧基甲卡西酮-D3、3,4-亚甲二氧基甲基苯丙胺-D5、甲氧麻黄酮-D3、去甲氯胺酮-D4、氯胺酮-D4、苯甲酰爱康宁-D3、利他林-D9、曲马多-D3、可卡因-D3、芬太尼-D5、丙烯酰芬太尼-D5、呋喃芬太尼-D5、异丁酰芬太尼-13C6、2-亚乙基-1,5-二甲基-3,3-二苯基吡络烷-D3、美沙酮-D3、戊酰基芬太尼-D5、安眠酮-D7、去甲乙酰芬太尼-13C6、去甲芬太尼-D5和右美沙芬-D3为E组。
45种待测化合物混合储备溶液:将A组、B组、C组的待测化合物按照预设的质量浓度使用甲醇稀释,混合,配制成45种待测化合物混合储备溶液,其中A组的质量浓度最高,质量浓度为1 μg/mL;B组质量浓度为A组的1/100(10 ng/mL);C组质量浓度为A组的1/10(100 ng/mL)。
38种内标化合物混合储备溶液:将D组、E组的内标化合物按照预设的质量浓度使用甲醇稀释,混合,配制为38种内标化合物混合储备溶液,其中D组的质量浓度为200 ng/mL;E组的质量浓度为2 ng/mL。
系列混合标准工作溶液:以甲醇-水(体积比为2∶8)为稀释剂,将上述两种化合物混合储备溶液配制成系列混合标准工作溶液,其中A组质量浓度分别为5、10、20、50、100 ng/mL,B组质量浓度分别为50、100、200、500、1 000 pg/mL,C组质量浓度分别为0.5、1、2、5、10 ng/mL。内标D组质量浓度为20 ng/mL,内标E组质量浓度为内标组D的1/100,其质量浓度为0.2 ng/mL。
1.4 实验方法
依次用适量的水和丙酮振荡洗涤毛发两次,晾干后将毛发剪成约1 mm段,置于液氮中冷冻研磨成粉末,称取20 mg经粉碎的毛发样品,加入20 ng的内标化合物(以E组计)和1 mL的1 moL/L硫酸锌甲醇溶液(体积比为4∶6)超声提取30 min,于12 000 r/min转速下离心5 min,取出全部上清液分散至50 mL pH为6的纯水中,混匀后以2 mL/min通过HLB固相萃取柱,上样结束后加入10 mL超纯水淋洗柱子。抽干柱子,使用10 mL甲醇洗脱,收集洗脱液并浓缩至近干,加入200 μL的20%甲醇溶液复溶,混匀后采用液相色谱-串联质谱法检测。
2. 结果与讨论
2.1 固相萃取柱与pH的选择
考察了MCX和HLB 两种不同的固相萃取柱,在pH分别为2、4、6时对45种待测化合物回收率的影响。甲醇超声提取毛发30 min后,离心取出上清液,往上清液中加入20 ng(以A组计)的目标化合物和20 ng(以E组计)的内标化合物。分散至50 mL的pH分别为2、4和6的纯水中,摇匀,经固相萃取得到待测样品溶液。不同pH下的萃取效果如图1所示,从图1中可以看出,受毛发的基质影响,在不同pH值的条件下,采用MCX柱萃取时,部分物质(如乙酰芬太尼、奥芬太尼和芬太尼)无法实现定量回收。而使用HLB固相萃取柱也有类似的情况,仅在pH为6时,HLB柱可实现所有待测物的定量回收,待测化合物的加标回收率为60%~140%,提取效果较好,因此选择HLB柱作为固相萃取柱,pH为6。
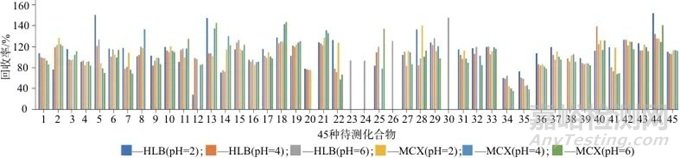
图1 MCX与HLB固相萃取柱在pH为2、4、6条件下的回收率
Fig. 1 Recoveries of MCX and HLB solid phase extraction column at pH 2, 4, 6
1—吗啡;2—卡西酮;3—甲卡西酮;4—苯丙胺;5—苄基哌嗪;6—甲基苯丙胺;7—乙卡西酮;8—3,4-亚甲二氧基甲卡西酮;9—可待因;10—3,4-亚甲基二氧基苯丙胺;11—3,4-亚甲二氧基甲基苯丙胺;12—甲氧麻黄酮;13—对甲氧基甲基苯丙胺;14—O6-乙酰吗啡;15—去甲氯胺酮;16—氯胺酮;17—苯甲酰爱康宁;18—利他林;19—曲马多;20—海洛因;21—可卡因;22—亚甲基二氧吡咯戊酮;23—乙酰芬太尼;24—奥芬太尼;25—丙烯酰芬太尼;26—芬太尼;27—呋喃芬太尼;28—异丁酰芬太尼;29—2-亚乙基-1,5-二甲基-3,3-二苯基吡络烷;30—4-氟丁酰芬太尼;31—美沙酮;32—戊酰基芬太尼;33—安眠酮;34—硝甲西泮;35—氟硝西泮;36—地西泮;37—阿普唑仑;38—四氢大麻酸;39—三唑仑;40—去甲乙酰芬太尼;41—去甲芬太尼;42—艾司唑仑;43—氯硝西泮;44—右美沙芬;45—哌替啶
另外考察了500 mg(6 mL)和200 mg(6 mL) 2种不同规格的HLB固相萃取柱对45种待测化合物回收率的影响。结果表明,使用500 mg(6 mL)的柱子并没有改善上述情况下的萃取效果,反而某些化合物还出现了基质增强的现象,如芬太尼类化合物、MDA、O6-乙酰吗啡和四氢大麻酸等回收率增加了20%~80%,可能是由于500 mg(6 mL)的柱子吸附了更多的毛发中的蛋白,在洗脱步骤被一并带入到洗脱液中,导致了更强的基质效应,因此选择HLB柱的规格为200 mg(6 mL)。
2.2 提取方法的选择
考察了酸性水解、甲醇超声提取与碱性水解对45种待测化合物回收率的影响。其中酸性水解的方法为将粉碎后的毛发样品20 mg置于1 mL 0.1 mol/L盐酸溶液中12 h,离心后取出上清液并分散于纯水中回调pH为6,其余步骤与1.4实验方法相同。碱性水解的方法为将粉碎后的毛发20 mg置于1 mL 1 mol/L NaOH溶液中,在60 ℃水解1 h,离心后取出上清液并分散于纯水中回调pH为6,其余步骤与1.4中实验方法相同。甲醇超声提取的方法为将粉碎后的毛发20 mg置于1 mL甲醇中超声30 min,离心后取出上清液并分散于纯水中调节pH为6,其余步骤与1.4中实验方法相同。
不同提取溶剂对毛发中的毒品的提取效果如图2所示,从图2中可以看出,采用碱性水解的方式提取毛发的效果最差,可能是在碱性条件下毛发溶解,毛发中的蛋白、色素等杂质进入提取液中,影响了固相萃取的吸附效果[18]。酸性水解的结果与甲醇超声提取的效果较为接近,但是对甲氧基甲基苯丙胺、利他林和异丁酰芬太尼的提取效率较差,鉴于酸性水解萃取时间长和操作步骤繁琐,为提高提取效率,最终选取甲醇超声提取作为提取方式。
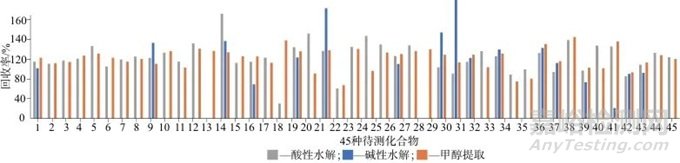
图2 不同提取溶剂对毛发中的毒品的回收率
Fig. 2 Recoveries of drugs in hair by different extraction solvents
1—吗啡;2—卡西酮;3—甲卡西酮;4—苯丙胺;5—苄基哌嗪;6—甲基苯丙胺;7—乙卡西酮;8—3,4-亚甲二氧基甲卡西酮;9—可待因;10—3,4-亚甲基二氧基苯丙胺;11—3,4-亚甲二氧基甲基苯丙胺;12—甲氧麻黄酮;13—对甲氧基甲基苯丙胺;14—O6-乙酰吗啡;15—去甲氯胺酮;16—氯胺酮;17—苯甲酰爱康宁;18—利他林;19—曲马多;20—海洛因;21—可卡因;22—亚甲基二氧吡咯戊酮;23—乙酰芬太尼;24—奥芬太尼;25—丙烯酰芬太尼;26—芬太尼;27—呋喃芬太尼;28—异丁酰芬太尼;29—2-亚乙基-1,5-二甲基-3,3-二苯基吡络烷;30—4-氟丁酰芬太尼;31—美沙酮;32—戊酰基芬太尼;33—安眠酮;34—硝甲西泮;35—氟硝西泮;36—地西泮;37—阿普唑仑;38—四氢大麻酸;39—三唑仑;40—去甲乙酰芬太尼;41—去甲芬太尼;42—艾司唑仑;43—氯硝西泮;44—右美沙芬;45—哌替啶
2.3 蛋白沉淀剂的影响
考察了沉淀蛋白剂对45种待测化合物的回收率影响。硫酸锌是常见的蛋白沉淀剂,常见于食品分析检测时沉淀蛋白,主要原理是溶液中的Zn2+与蛋白结合产生沉淀,具有良好的除蛋白效果,因此使用1 mol/L硫酸锌甲醇溶液(体积比为4∶6)替代甲醇对毛发进行超声提取,在加标量为20 ng(以吗啡计)的条件下45种目标物回收率为73.4%~123.5%,回收率良好,因此使用硫酸锌甲醇溶液进行超声提取。此外,考察了硫酸锌甲醇溶液对萃取效果的影响,结果表明,蛋白沉淀剂硫酸锌溶液甲醇的加入提高了抗干扰性,降低了样品中基质效应,使制备的样品更洁净,可以减少仪器的维护次数。
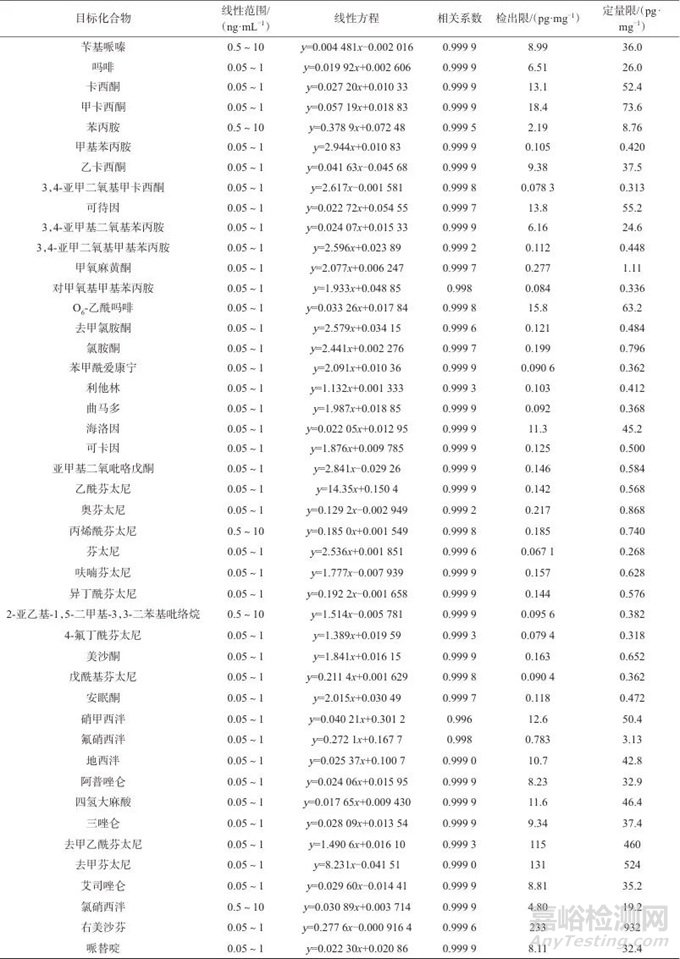
2.4 线性方程与检出限
在1.2仪器条件下,对系列混合标准工作溶液进行测定,以目标化合物的质量浓度(x)为横坐标,以化合物的峰面积与对应内标的峰面积的比值(y)为纵坐标,内标法进行定量分析。取7份经冷冻粉碎研磨的阴性毛发粉末样品,加入一定量的目标化合物,经处理后,计算7次测定结果的标准偏差。以3倍标准偏差作为方法检出限,以10倍标准偏差作为方法定量限。45种毒品及代谢物的线性范围、线性方程、相关系数、检出限及定量限见表3。由表3可知,45种毒品及其代谢物的质量浓度在各自范围内与色谱峰面积线性关系良好,相关系数均不小于0.996,检出限为0.067 1~233 pg/mg,与标准SF/Z JD0107025—2018相比,该方法中吗啡、苯丙胺、甲基苯丙胺等物质的检出限低了两个数量级,灵敏度更高。
表3 45种毒品及代谢物的线性范围、线性方程、相关系数、检出限及定量限
Tab. 3 Linear ranges, linear equations, correlation coefficients, detection limits and quantitation limits of 45 drugs and metabolites
2.5 加标回收与精密度试验
往经冷冻粉碎研磨的阴性毛发粉末中按低、中、高3个浓度水平加入目标化合物,每一个加标浓度平行制备6份样品溶液,在1.2仪器工作条件下分别进行测定,加标回收及精密度试验结果见表4。
表4 加标回收与精密度试验结果
Tab. 4 Results of standard recoveries and precision test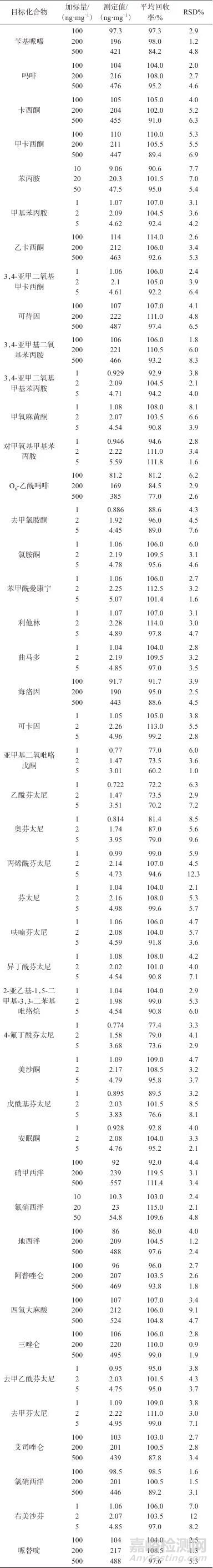
由表4可知,3种不同加标浓度下,45种毒品及其代谢物的平均回收率为60.2~119.5%,测定结果的相对标准偏差(RSD)为0.9%~12.3%(n=6)。该方法准确度与精密度较好,与文献[15-19]相比,该方法可同时分析的毒品种类更多,且消耗的毛发用量更低。
3. 结语
建立了固相萃取-液相色谱-串联质谱法测定毛发中45种毒品及代谢物。对比了提取方式、pH值和固相萃取柱种类等对45种毒品及其代谢物回收率的影响。使用硫酸锌甲醇溶液替代甲醇作为提取剂,能有效减少因提取毛发时引入的蛋白质所产生的基质效应,同时减少蛋白质对分析系统的污染。与此同时,通过使用固相萃取技术,对硫酸锌甲醇提取液进一步净化,减少样品中出现假阳性的几率。与现有的国家标准相比,该方法具有同时测定的物质种类多、抗干扰能力强的优点,为毒品司法鉴定检测提供了一种可靠、科学、高效的手段。
参考文献:
1 汪予佳,汪孝匡,方圆,等.毒品检测技术应用研究进展[J].中国药物滥用防治杂志,2023,29(8):1 303.
WANG Yujia,WANG Xiaokuang,FANG Yuan,et al. Research progress on the application of drug detection technology[J]. Chinese Journal of Drug Abuse Prevention and Treatmen,2023,29(8):1 303.
2 丁艳,刘培培,闻武,等.环境中毒品检测技术研究进展与展望[J].中国法医学杂志,2024,39(1):14.
DING Yan,LIU Peipei,WEN Wu,et al. Research progress and prospect of drug detection technology in the environment [J]. Chinese Journal of Forensic Medicine,2024,39(1):14.
3 方正,詹燮峰,陈银彬,等.毒品检测方法的技术分类[J].中国药物滥用防治杂志,2024,30(1):5.
FANG Zheng,ZHAN Sifeng,CHEN Yinbin,et al. Technical classification of drug detection methods[J]. Chinese Journal of Drug Abuse Prevention and Control,2024,30(1):5.
4 张婷婷,吴健美,乔宏伟,等.毛发中毒品检测技术及其在毒情监测中的应用[J].警察技术,2022(5):19.
ZHANG Tingting,WU Jianmei,QIAO Hongwei,et al. Drug detection technology in hair and its application in drug monitoring [J]. Police Technology,2022(5):19.
5 KEMPSON I M, LOMBI E. Hair analysis as a biomonitor for toxicology, disease and health status[J]. Chemical Society Reviews, 2011,40(7):3 915.
6 李利华,万敬伟,孔维刚.气相色谱质谱(GC/MS)检验新型毒品依托咪酯[J].铁道警察学院学报,2024,34(1):101
LI Lihua,WAN Jingwei,KONG Weigang. Gas chromatographic mass spectrometry (GC/MS) to detect new drug etomide [J]. Journal of Railway Police Academy,2024,34(1):101
7 努尔艾力·塔依尔,毛新民,车文森,等. GC、GC-MS法定性定量法检测血液和尿液中阿片类毒品及其代谢物的含量[J].新疆医科大学学报,2016,39(7):849
NUERAILI Tayier,MAO Xinmin,CHE Wensen,et al. GC and GC-MS statutory quantitative method to detect the content of opioids and their metabolites in blood and urine [J]. Journal of Xinjiang Medical University,2016,39(7):849
8 胡骏杰,郝敬梅,刘飞,等.超高效液相色谱-串联质谱结合内标法快速检测毛发中15种毒品及代谢物[J].分析科学学报,2021,37(3):287
HU Junjie,HAO Jingmei,LIU Fei,et al. Ultra high performance liquid chromatography-tandem mass spectrometry combined with internal standard method to quickly detect 15 drugs and metabolites in hair [J]. Journal of Analytical Science,2021,37(3):287
9 吴君金,张静红,刘遥,等.毛发中9种毒品的LC-MS/MS定性定量分析方法研究[J].化纤与纺织技术,2022,51(11):32.
WU Junjin,ZHANG Jinghong,LIU Yao,et al. Research on the qualitative and quantitative analysis method of LC-MS/MS of 9 drugs in hair [J]. Chemical fiber and textile technology,2022,51(11):32
10 杨乔,吴波,田冰冰,等. QTRAPLC-MS/MS同时测定毛发中20种毒品及代谢物[J].中国法医学杂志,2020,35(3):296.
YANG Qiao,WU Bo,TIAN Bingbing,et al. Simultaneous determination of 20 drugs and metabolites in hair using QTRAP LC-MS/MS[J]. Chinese Journal of Forensic Medicine,2020,35(3):296.
11 TURKMEN Z, ARSLAN Z, OKA M, et al. LC-MS/MS quantification of olanzapine in hair after alkaline digestion[J]. Drug Testing and Analysis,2024.
12 LEE S, CHOI B, KIM J, et al. An LC-MS/MS method for the determination of five erectile dysfunction drugs and their selected metabolites in hair[J]. Journal of Chromatography B, 2015, 978:1.
13 侯峰,杨崇俊,张建强,等.毛发中8种常见精神类药物的LC-MS/MS检测方法[J].中国药物依赖性杂志,2019,28(6):441.
HOU Feng,YANG Chongjun,ZHANG Jianqiang,et al. LC-MS/MS detection methods for 8 common psychotropic drugs in hair [J]. Chinese Journal of Drug Dependence,2019,28(6):441.
14 吴君金,张静红,刘遥,等.毛发中9种毒品的LC-MS/MS定性定量分析方法研究[J].化纤与纺织技术,2022,51(11):32
WU Junjin,ZHANG Jinghong,LIU Yao,et al. Research on the qualitative and quantitative analysis method of LC-MS/MS of 9 drugs in hair [J]. Chemical fiber and textile technology,2022,51(11):32
15 苏佳丽. UPLC-MS/MS同时检测人体毛发中26种滥用药物[J]. 中国法医学杂志,2022,37(5):490.
SU Jiali. UPLC-MS/MS simultaneously detects 26 drugs and their metabolites in human hair [D]. Chinese Journal of Forensic Medicine,2022,37(5):490.
16 谢普,王铁杰,殷果,等. UPLC-MS/MS法测定吸毒者头发中10种毒品代谢物的含量[J].药物分析杂志,2014,34(3):516.
XIE Pu,WANG Tiejie,YIN Guo,et al. UPLC-MS/MS method to determine the content of 10 drug metabolites in the hair of drug addicts [J]. Journal of Drug Analysis,2014,34(3):516.
17 王铁杰,詹华强,孙新珺,等. UPLC-MS/MS同时测定头发中十种残留毒品含量——深港创新合作共建头发验毒服务平台[J].中国药物滥用防治杂志,2012,18(6):317.
WANG Tiejie,ZHAN Huaqiang,SUN Xinjun,et al. UPLC-MS/MS simultaneously determines the content of ten residual drugs in hair-Shenzhen-Hong Kong innovative cooperation to build a hair drug testing service platform [J]. Chinese Journal of Drug Abuse Prevention and Control,2012,18(6):317.
18 陈顺琴,唐美,黄江,等.超声提取-超高效液相色谱-串联质谱法测定毛发中常见毒品及代谢物的含量[J].理化检验(化学分册),2022,58(1):58.
CHEN Shunqin,TANG Mei,HUANG Jiang,et al. Determination of common drugs and metabolites in hair by ultrasonic extraction-ultra ultra high performance liquid chromatography-tandenm mass spectrometry [J]. Physical Testing and Chemical Analysis (Part B:Chemical Analysis),2022,58(1):58.
19 王伟,刘国如,徐唯哲,等.人毛发中14种常见毒品及其主要代谢物的UPLC-MS/MS分段离子切换法测定[J].国际药学研究杂志,2020,47(4):304.
WANG Wei,LIU Guoru,XU Weizhe,et al. Determination of 14 common drugs and their main metabolites in human hair by UPLC-MS/MS segmented ion switching method [J]. International Journal of Pharmaceutical Research,2020,47(4):304.
引用本文: 赵嘉辉,彭诗琪,赖华杰,等. 固相萃取-液相色谱-串联质谱法测定毛发中45种毒品及代谢物[J]. 化学分析计量,2024,33(11):64. (ZHAO Jiahui, PENG Shiqi, LAI Huajie, et al. Determination of 45 drugs and their metabolites in hair by liquid chromatography-tandem mass spectrometry with solid phase extraction coupled[J]. Chemical Analysis and Meterage, 2024, 33(11): 63.)

来源:化学分析计量


