摘要
国际人用药品注册技术协调会( ICH) Q11是原料药开发和生产的技术指导原则,系统阐述了原料药工艺开发的基本方法和起始原料的选择原则; ICH Q11问答文件进一步阐述了选择起始原料的具体要求。本文参考 ICH Q11及问答文件的要求,围绕创新药药学研究的阶段性规律,讨论化学创新药技术审评中基于 ICH Q11的考虑。
引言
国际人用药品注册技术协调会( ICH) Q11 是原料药的开发和生产( 化学实体和生物技术/生物实体药物) 技术指导原则,2012 年 5 月正式实施,主要内容是指导原料药( 包括化学实体和生物技术/生物实体药物) 的开发和生产,包括生产工艺开发、工艺过程控制策略、起始原料的选择、生命周期管理等方面,系统阐述了原料药工艺开发的基本方法和选择起始原料的原则,导通用技术文件( the common technical document,CTD) 模块“3.2.S.2.2”至“3.2.S.2.6”内容的整理与撰写[1]。同时,为方便业界更好地理解 ICH Q11 的技术要求,ICH 制定了关于 Q11 的问答文件( 原料药的开发和生产 Q&A) ,2017 年 8 月正式实施,以问答的形式阐述了指定起始原料的具体要求,明确选择起始原料的部分共性问题,引入起始原料选择的决策树[2]。国家药品监督管理局于 2020 年 1 月 21 日发布公告,推荐药品注册申请人按照 ICH Q11 及问答文件的要求开展相关研究[3],国家药品监督管理局药品审评中心也于 2019 年发布了 ICH Q11 以及问答文件的中文翻译版[4]。
ICH Q11 适用于 ICH Q6A 与 Q6B 描述的新原料药[5] /原液,与监管机构充分沟通交流后,也可用于其他类型的产品[1]。目前在化学药品1~5类注册申请原料药审评中,均执行 ICH Q11。化学药品1类为境内外均未上市的新的结构明确的、具有药理作用的化合物,且具有临床价值的药品[6],ICH Q11正式实施后,遵循 ICH Q11 的基本原则开展化学创新药技术审评。本文结合笔者创新药审评工作实践,重点讨论化学创新药技术审评中基于 ICH Q11的考虑。
一、创新药原料药起始原料选择和质量控制要求
1.1 起始原料的选择具有阶段性
创新药药学研究的广度和深度伴随临床试验的进展推进,不同研究阶段的药学研究目的不同,药学技术审评要求与研发阶段相适应。
起始原料是用于生产原料药并成为该原料药结构的重要组成部分的一种原料、中间体或其他原料药,通常应具有明确的理化性质和结构[7]。创新药起始原料的选择与质量控制具有阶段性与渐进性的特点。
申请早期临床试验阶段,对原料药理化性质与制备工艺的认知有限,建立了初步的起始原料质量控制方法和限度,临床试验样品杂质水平不得超出动物安全性实验数据所支持的相应杂质的水平。对于起始原料杂质限度比较宽泛,制备临床试验原料药的起始原料杂质水平不应高于毒理实验批次原料药采用起始原料的杂质水平,验证起始原料杂质检查方法的专属性,加强起始原料的质量控制。
随着临床试验的进展,对原料药工艺研究及认知逐步丰富,杂质谱研究( 包括杂质在后续步骤的转化清除情况) 进一步深入,应参照 ICH Q11 要求研究和选择合理的起始原料。启动Ⅲ期临床试验前( 或支持新药上市的关键临床试验前) ,建议与药品审评机构沟通起始原料选择的合理性,Ⅲ期临床试验期间参照 ICH Q11 完善工艺研究与起始原料的质量控制。
纳入突破性治疗、拟申请附条件批准的创新药,临床试验期间应加强研究团队内部沟通和协调,根据临床试验进展及目标协调加快药学开发进度,并与药品审评机构及早进行沟通,参照 ICH Q11 研究和选择合理的起始原料。
1.2 基于 ICH Q11 以及问答文件原则进行研究和选择
创新药原料药起始原料主要基于 ICH Q11以及问答文件的以下原则进行研究和选择: ① 起始原料应当具备明确的化学特性和结构,作为重要的结构片段并入原料药的结构中。未分离的中间体通常不被考虑作为合适的起始原料。② CTD 模 块“3.2.S.2.2”应包含对原料药的杂质谱产生影响的生产步骤,充分描述原料药的生产工艺,明确杂质在工艺过程中的产生、去向与清除,控制策略满足杂质控制要求。③ 采用汇聚型原料药生产工艺的每个分支开始于 1 个或多个起始原料。每个分支中从首次使用起始原料的步骤开始,需要遵循生产质量管理规范,并结合适当的控制策略为原料药的质量提供保证。④ 在选择起始原料时应当考虑上述全部原则,而不是孤立地严格遵循每一个原则。
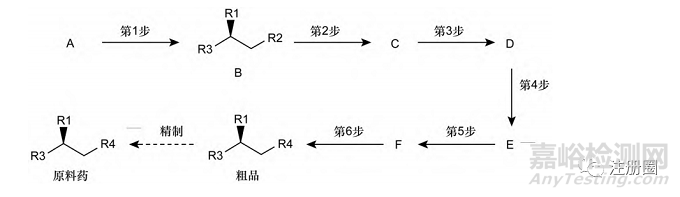
ICH Q11 通过案例进一步阐述如何选择起始原料,工艺路线见图 1,假定第1步仅对原料药的对映异构体水平产生影响,原料药手性结构由第1步产物化合物 B 引入,手性中心的立体构型在后续步骤未发生转变; 第 2 步与第 3 步对原料药杂质谱无影响,原料药中的主要杂质均在第 4 步至第 6 步产生;基于各工艺步骤对原料药杂质谱产生的影响,如果化合物 D 的对映异构体能够有效控制,同时满足起始原料选择的其他原则,化合物 D 可以作为起始原料。
▲ 图1-ICH Q11 起始原料选择案例
1.3 重点关注问题
结合ICH Q11 选择起始原料的案例,可以看出,明晰杂质在原料药生产工艺中的转化清除途径,建立有效的杂质控制策略,是合理选择起始原料的重要基础。不同结构原料药的生产工艺复杂程度不同,需结合原料药杂质控制策略,参照ICH Q11 及问答文件选择合理的起始原料。
起始原料不应对原料药的质量控制( 特别是杂质水平) 产生重大风险,起始原料引入的杂质( 包括有关物质、致突变杂质、毒性试剂/溶剂、金属催化剂等) 及其在原料药工艺中的转化产物能够得到有效控制。充分研究原料药潜在杂质产生来源以及在工艺中的转化与清除,制定合理的工艺过程控制策略( 包括工艺参数、物料/中间体的控制) ,参照 ICH Q11 及问答文件综合论证起始原料选择的合理性[8]。
根据原料药质量控制的需要制定起始原料内控质量标准,结合原料药生产工艺对杂质的清除能力制定起始原料的已知与未知单个杂质限度,杂质检查方法能有效分离并检出相关杂质; 加强起始原料生产企业的质量审计,起始原料生产企业能够持续稳定地生产符合要求的起始原料。
半合成原料药是通过将生物来源的成分( 如微生物发酵或植物中提取的成分) 与化学合成方法相结合得到的原料药,ICH Q11 也阐述了半合成原料药起始原料选择的原则: ① 通常应当从源物质( 微生物或植物) 开始描述生产工艺。② 如果能够证明合成工艺中的一个已分离的中间体符合 Q11 对化学合成原料药起始原料的选择原则,该分离中间体可作为起始原料,但应当对所选择的起始原料的质量控制进行特别评估,包括潜在杂质、发酵过程/植物提取过程对原料药杂质谱的影响,还应该论述来源于微生物和其他污染物的安全风险。
微生物发酵工艺过程控制复杂,工艺参数的微小变化可能导致产品组分比例与杂质水平的显著变化。对于半合成原料药,除了将微生物发酵与分离纯化纳入原料药工艺,还应当将其纳入原料药企业的生产质量管理体系,保证原料药质量持续稳定。对于结构相对简单( 如氨基酸) 或具有较长生产历史、已在多种原料药的生产工艺中使用、质量属性已认知充分的发酵制备物料( 如 6-APA,7-ACA) ,在保证质量控制满足要求的前提下,可以参照 ICH Q11 作为起始原料。此外,通过微生物分类学与基因测序方法鉴定发酵菌种的种属,关注菌种传代可能发生的回复突变; 固定碳源、氮源等发酵物料的来源,防止储存期间霉变,评估产生黄曲霉素的风险并制定控制措施。
二、创新药原料药工艺开发及过程控制
创新药的工艺研究与控制同样具有阶段性的特点,对药物的关键质量属性( critical quality assurance,CQA) 和工艺过程控制的理解伴随临床试验的进展而逐渐深入,原料药生产过程控制要求逐步加强。
在确认目标质量概况 ( quality target product profile,QTPP) 的 基 础 上,鼓励遵循质量源于设计( quality by design,QbD) 的理念,通过实验设计( design of experiment,DoE) 加深工艺认知,同时重视传统的单因素实验确定关键工艺因素的重要性,在确定影响原料药质量属性的工艺因素基础上,通过实验设计确定参数最佳控制范围。伴随临床试验的进展,加深原料药关键质量属性的理解,识别影响原料药质量的工艺参数,优化工艺过程控制。
参照 ICH Q11及问答文件选择确定起始原料后,参照 ICH Q11 关于原料药开发与过程控制策略的原则继续优化工艺参数,对工艺与批量变更开展针对性研究,确定商业规模批量生产下的关键工艺参数控制范围,通过工艺验证进一步确认生产工艺的持续稳定,在申请上市前建立稳定、持续、可重现的工业化生产工艺,构建完善的药品质量控制体系。
三、重视创新药临床试验期间的沟通交流与药学研究
3.1 及时进行药学沟通交流( 包括Ⅲ期临床试验前沟通交流)
笔者整理美国 FDA 公开的 2010—2021 年批准上市的 210 个创新化学药品Ⅲ期临床
试验前( end of phase II,EoP2) 与上市申请前( prenew drug application,pre-NDA) 的药学沟通交流信息[9],讨论的共性问题包括起始原料的选择、杂质控制、溶出度方法区分力、稳定性研究等,其中 42 个创新药( 不完全统计) 针对起始原料选择的合理性进行讨论。最近我国申请上市的 1 类化学药中,未及时进行药学方面的沟通交流,尤其是Ⅲ期临床试验前未及时申请进行药学沟通交流是存在的主要问题,最近 3 年在我国提出的 40 件Ⅲ期临床试验前药学沟通交流中,仅 12 件针对起始原料选择的合理性进行讨论。
部分申请人在新药申请上市前与药品审评机构沟通交流时,才发现起始原料的选择和质量控制存在问题,合成路线需要前延,需要重新寻找符合要求的生产企业和进行工艺转移,影响了创新药的注册申请进度。
原料药工艺研究与验证的周期通常较长,如果在临床试验关键阶段甚至申请上市前进行合成路线前延,大量的工艺研究与验证势必推迟药品申请上市的进程,因此建议申请人在关键临床试验启动前参照 ICH Q11 及问答文件合理选择起始原料,及时与审评机构沟通,结合对生产工艺的理解合理确定《药品生产质量管理规范》( Good ManufacturingPractice of Medical Products,GMP) 条件下的生产步骤,尽早确定起始原料; 申请上市前,结合 ICH Q11及问答文件充分论证起始原料选择的合理性,申请上市前与审评机构沟通交流达成共识。
3.2 遵循 ICH Q11深入开展研究
笔者在创新药上市申请的药学审评工作中,发现大多数原料药的起始原料与工艺控制存在较多问题,体现在起始原料选择不合理、杂质控制宽泛、工艺参数依据不足等方面。需要参照 ICH Q11 与问答文件全面系统开展原料药工艺开发,合理选择起始原料,根据杂质( 包括致突变杂质) 在工艺中的转化清除研究制定起始原料与中间体的杂质控制策略[10]; 确认原料药关键质量属性,深入研究工艺参数对产品质量的影响,为工艺参数控制范围的制定提供充分依据,申请上市前在商业化产地完成工艺验证。
四、总结
ICH Q11 与问答文件的基本原则已应用于我国化学创新药的技术审评。本文从 ICH Q11 与问答文件的原则要求出发,讨论基于 ICH Q11 及问答文件开展化学创新药起始原料的选择、原料药工艺开发技术审评的一般考虑,为化学创新药的技术审评提供参考,同时为化学创新药的药学研究存在的问题提出建议。
参考文献
[1] ICH. Development and manufacture of drug substances[EB/OL]. ( 2012 - 05 - 01) [2021 - 12 - 18]. https: / /database.ich. org /sites/default /files/Q11%20Guideline. pdf.
[2] ICH. Development and manufacture of drug substances Questions& Answers[EB /OL]. ( 2017 - 08 - 23) [2021 - 12 - 18]. https: / /database. ich. org /sites/default /files/Q11 _Q% 26As_Q%26As. pdf.
[3] 国家药品监督管理局. 国家药监局关于推荐适用《Q8( R2) : 药品研发》等4 个国际人用药品注册技术协调会指导原则的公告[EB/OL]. ( 2021 -01 -21) [2021 -12 -19]. https: / /www. nmpa.gov. cn /yaopin /ypggtg /ypqtgg /20200121171001817. html.
[4] 国家药品监督管理局药品审评中心. Q11 原料药开发和生产( 化学实体和生物技术生物实体药物) ( 中文翻译公开征求意见稿) [EB/OL]. ( 2012 - 05 - 01) [2021 - 12 - 19]. https: / /www. cde. org. cn /ichWeb /guideIch /downloadAtt /2 /6ea3d10b2be92c29e5e0214801eeae25.
[5] ICH. Specifications test procedures and acceptance criteria for new drug substances and new drug products chemical substances[EB/OL]. ( 1999 - 10 - 06) [2022 - 01 - 13]. https: / /database. ich. org /sites/default/files/Q6A%20Guideline. pdf.
[6] 国家药品监督管理局. 国家药监局关于发布化学药品注册分类及申报资料要求的通告[EB/OL]. ( 2020 - 06 - 30) [2022 -01 - 10]. https: / /www. nmpa.gov. cn /xxgk /ggtg /qtggtg /20200630180301525. html.
[7] ICH. Good manufacturing practice guide for active pharmaceuti cal ingredients[EB/OL]. ( 2000 - 11 - 10) [2022 - 01 - 19].https: / /database. ich. org /sites/default /files/Q7% 20Guideline.pdf.
[8] FAUL MM,ARGENTINE,EGAN M,et al. Part 3: designation and justification of API starting materials: proposed framework for alignment from an industry perspective[J]. Org Process Res Dev,2015,19( 8) : 915 - 924.
[9] FDA. Drugs@ FDA[EB /OL].[2022 - 01 - 19]. https: / /www.accessdata. fda. gov /scripts/cder/daf /.
[10] 王云,朱建伟. 化学合成原料药起始物料的选择原则[J]. 中国医药工业杂志,2022,53( 5) : 728 - 734.