您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2025-01-13 12:41
① FDA医疗器械分类有哪些判定途径?
· 常规途径:FDA具有多种途径提供医疗器械分类信息,可通过产品分类数据库、510(k)数据库、PMA数据库、De Novo数据库、510(k)豁免的I类和II类器械指南文件库、同工业与消费者教育部(DICE)沟通、同组合产品办公室沟通、生物制品与研究中心(CBER)监管的特定器械信息等预先评估确认产品分类。
· 513(g)请求途径:若通过常规途径无法判定分类,则可以进行513(g)分类请求以确认器械分类及对应的法规要求。
② 513(g)分类请求不适用哪些情况?
· FDA不负责实质等效性、安全性及有效性相关数据的审查。
· 不回复涉及支持上市申请许可或批准的非临床、动物或临床测试的问题,此类问题可向FDA文件控制中心提交Q-submission资料由相应部门审查并回复。
· 对513(g)请求的回复,既不是器械分类决策,也不构成FDA的审查批准或上市批准,分类决策、上市许可或批准应当根据《联邦食品、药品和化妆品法案》(以下简称FD&C 法案)不同章节要求提交申报资料。
③ 513(g)分类请求需要提供什么信息?
1. 器械基本信息:请求者需要提供器械描述和功能,以便FDA评估其是否符合"器械"的定义。
2. 器械分类信息:请求者应提供器械类型和预期用途,以帮助FDA确定器械分类(例如分类法规)。
3. 相关法规要求:请求者需要说明器械可能适用的法规要求,包括是否需要提交PMA(预市场批准)或510(k)(预市场通知)。
4. 其他适用要求:如器械是否涉及辐射发射产品等其他FDA要求。
④ 513(g)分类请求应该如何提交?
可通过FDA线上提交系统以eCopy或eSTAR 的方式提交请求文件。
⑤ 513(g)分类请求需要付费吗?
根据FD&C法案第738(a)(2)(A)(ix)条款,FDA要求对513(g)信息请求向用户收取费用,FDA在收到所有费用后开始审查请求。
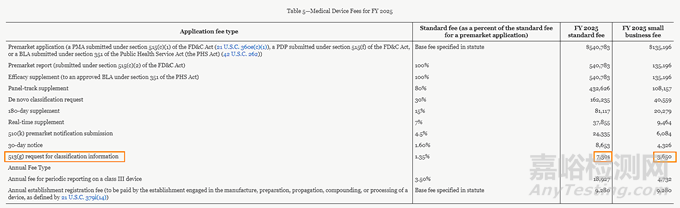
2025年(2024年10月1日开始执行)的513(g)分类请求评估费是7301美金,小企业申请费用减免后评估费用为3650美金。
⑥ 513(g)分类请求会有哪些结果?
1. 属于“医疗器械”范畴:
· 若为未分类的FD&C法案修订前器械类型,受510(k)要求约束;
· 若为尚未重新分类的FD&C法案修订后器械类型,受上市前批准要求约束;
· 已有分类的器械类型,FDA会识别适用的分类法规、器械类别(如I、II或III类)、上市申请途径(需符合510(k)要求的I类或II类、可豁免510(k)要求的I类或II类、需符合510(k)或PMA要求的III类等)。
2. 不属于“医疗器械”范畴:
· 可能是FDA监管的另一种产品,机构会提供FDA内另一相关部门的联系信息;
· 或许不是FDA拥有管辖权的产品;
· 管辖权不明的组合产品:建议联系组合产品办公室进一步讨论产品分配。若请求文件不完整,FDA无法提供相关信息时会联系提交者要求其补充,若30天内未收到回复可能考虑撤回513(g)。

来源:久顺集团技术服务


