您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2020-08-27 22:19
2019年9月13日,FDA发布指南《特殊510(k)项目(The Special 510(k) Program)》,该指南与同时发布的《简化510(k)项目(The Abbreviated 510(k) Program)》指南共同取代了1998年《新510(k)模式–上市前公告实质等同证明的替代方法(The New 510(k) Paradigm – Alternate Approaches to Demonstrating Substantial Equivalence in Premarket Notifications)》指南。为反映特殊510(k)路径的更新内容,FDA同时更新《510(k)拒收政策(RTA,Refuse to Accept Policy for 510(k)s)》和《传统510(k)和简化510(k)格式》两份指南。FDA发布的四份指南中,《特殊510(k)项目》指南内容发生了显著的变化。
一、背景
美国制造商向FDA提出意见,认为虽然1998年指南中特殊510(k)路径意在帮助节省时间和资金,但实际效果未达预期,原因是较多的特殊510(k)申请在递交申请后被转为传统510(k),且转换程序和标准缺乏透明性(clarity)。
为解决以上问题,2018年9月28日,FDA发布《特殊510(k)项目(草稿)》指南,紧接着又宣布开展特殊510(k)试点项目(Special 510(k) Pilot Program),2018年10月1日后受理的特殊510(k)申请都将纳入该试点项目,以验证指南中特殊510(k)路径是否可增加通过特殊510(k)路径上市的产品数量,减少传统510(k)数量,节省FDA审评资源。
2019年9月13日,FDA发布《特殊510(k)项目》指南,其内容与1年前发布的草稿版指南无实质性变化,可见该试点计划实施较为成功。
二、特殊510(k)路径要求的变化
(一)适用条件的变化
新指南未发布前,对已上市510(k)产品的改进,若不影响器械的预期用途,且不改变器械基本科学技术,可通过特殊510(k)路径申请上市。在该适用条件下,任何影响器械预期用途的适应症或标签变化、有可能改变器械基本科学技术的变化都将被转换为传统510(k),从而按照传统510(k)路径要求准备申报资料。为了规避以上问题,使更多产品通过特殊510(k)路径上市,更新的指南明确不再以是否改变影响适用症或改变基本科学技术作为适用条件,而以是否存在公认成熟方法评价该变化、是否能以摘要或风险分析表格的形式审评性能数据为特殊510(k)的适用条件。
更新的特殊510(k)路径适用条件为:
1.制造商提交的特殊510(k)申请是基于其自身已有510(k)产品的变化;
2.当(评价该变化带来的影响)性能数据不是必需的,或当可以使用公认成熟(well-established)的方法进行评估时;
3.可以以摘要或风险分析表格的形式完成实质等同性能数据的审评工作。
若不符合以上三条要求,特殊510(k)将转为传统510(k)。
(二)新增两种不适用特殊510(k)的情形
在最新的指南中,FDA特别说明“生物类器械通常不适用于特殊510(k),因为不太可能存在评价其变化的公认成熟方法,也不太可能以摘要或风险分析的表格呈现性能数据”。
指南明确当变化涉及超过3个不同技术主题(例如生物相容性、灭菌、电磁相容性等)时,将不适用特殊510(k),因为FDA很难在30天(特殊510(k)的审评时限)里完成技术审评。
(三)增加公认成熟方法的解释
指南明确了公认成熟的方法包括已获批510(k)中的方法(Method)、方案(Protocol)和接受标准(Acceptance criteria),同时也包括FDA认可标准(Recognized standard)中的方法、已发表的被广泛认可的方法、已批准De Novo和PMA中已被接受的方法。FDA强调了公认成熟的方法包括已经认证的医疗器械研发工具(MDDT,medical device development tools),以及FDA指南中的方法。
当与公认成熟方法存在偏离时,FDA明确微小偏离可能是可接受的,但方案(Protocol)或接受标准(Acceptance criteria)的显著偏离会致使申报器械不再适用特殊510(k)。
(四)特殊510(k)路径决策图变化
因特殊510(k)适用条件发生变化,相应决策图也发生了变化,附件1为变化后决策图,附件2为变化前决策图。
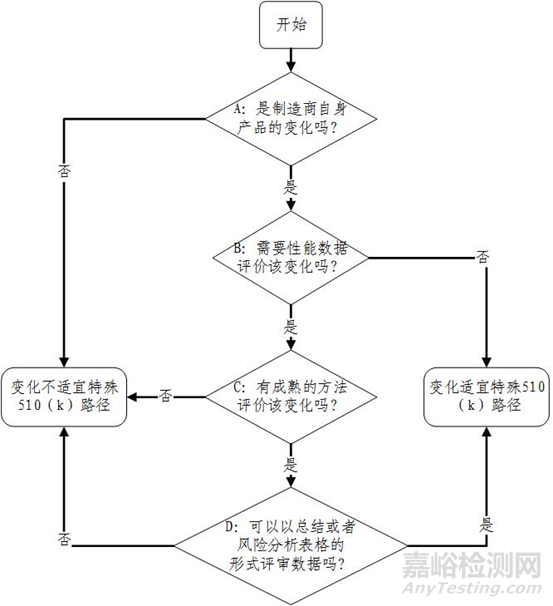
图1:2019年更新后特殊510(k)上市路径决策图。
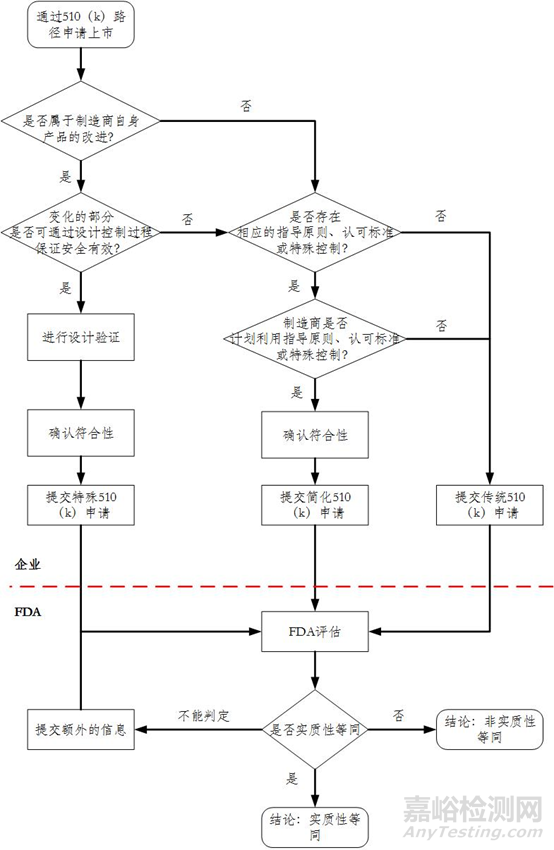
图2:1998年510(k)路径决策图,包括特殊510(k)。
三、510(k)拒收政策指南的变化
510(k)拒收政策指南最早于1993年发布,并于1994年、2012年、2015年、2018年、2019年2月数次更新,2019年9月13日发布的指南为目前最新版本。目前的拒收政策指南包括较多技术细节要求,例如“是否明确灭菌状态”“是否明确预期单次使用描述”“是否为药械组合”“WIFI、云技术相关要求”等。
该指南数次更新的原因是1993年和1994年版本指南内容属于行政相关的要求(administrative elements),未提出技术审评相关的要求。因此,较多存在明显技术缺陷的申报资料被予以受理,FDA审评员需投入大量时间撰写发补信以要求制造商提供开展实质审评所必须的全部信息。在一份研究报告中,FDA对2001-2010年间受理的510(k)申请中发出补全信息信申请所占的比例做了统计,发出补全信息信的比例随时间线性增高。2010年,FDA至少一次发出补全信息信的510(k)数量占全部受理510(k)数量的77%(见图3)。
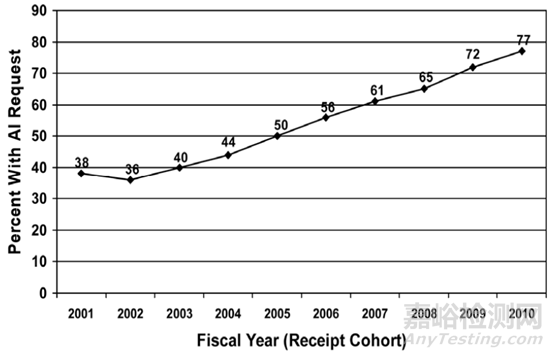
图3:FDA发出补全信息信(AI, Additional Information)的510(k)占全部受理510(k)数量的比例。
参考文献:
[1] The Special 510(k) Program - Guidance for Industry and Food and Drug Administration Staff
[2] Format for Traditional and Abbreviated 510(k)s - Guidance for Industry and FDA Staff
[3] The Abbreviated 510(k) Program - Guidance for Industry and Food and Drug Administration Staff
[4] Refuse to Accept Policy for 510(k)s - Guidance for Industry and Food and Drug Administration Staff.
[5] Analysis of Premarket Review Times under The 510(K) Program.

来源:未知


