您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2024-08-07 09:14
医疗器械的设计、制造工艺或监管和标准要求的变化将不可避免地发生,如果是一家初创公司,这些变化可能比成熟的企业来说更加频繁,在这两种情况下,需要妥善管理这些变化。那么这就意味着需要制定适当的流程和程序来确保:
* 一个正式的受控流程来请求变更
* 对请求的变更进行必要的审查和批准
* 管理和记录发生的变更
根据 ISO 13485:2016 和 FDA 的要求,对医疗器械的设计变更进行记录和控制也必须进行记录。如果更改不当,可能会导致重大问题或变得更糟,从而可能发生重大的产品质量和安全问题。
在此,将再次分享变更控制和变更管理流程所需的步骤,这些流程的有效实施将有效地促进变更的控制和记录,并满足所需的法规和标准要求。
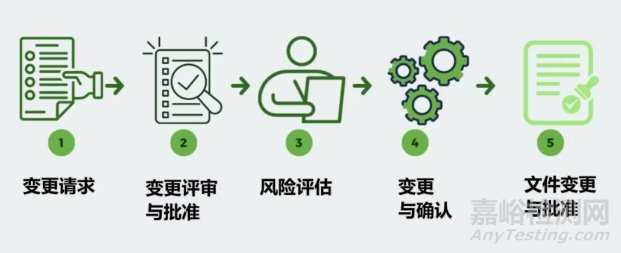
在我们查看上图所示的每个过程步骤之前,我们回顾一下 ISO 和 FDA 的要求:
1)ISO 13485:2016:7.3.9 设计和开发变更的控制
The organization shall document procedures to control design and development changes. The organization shall determine the significance of the change to function, performance, usability, safety and applicable regulatory requirements for the medical device and its intended use.
组织应将控制设计和开发更改的程序形成文件。组织应确定更改对于医疗器械功能、性能、可用性、安全、适用的法规要求及其预期用途的重要程度。
应识别设计和开发的更改。更改在实施前应经:
a)评审;
b)验证;
c)适当时,确认;
d)批准。
设计和开发更改的评审应包括评价更改对在制的或已交付的组成部件和产品的影响,以及对风险管理的输入或输出和产品实现过程的影响。
应保留更改及其评审和任何必要的措施的记录(见4.2.5)。
2)FDA 21 CFR Part 820.30(i) 设计变更
Each manufacturer shall establish and maintain procedures for the identification, documentation, validation or where appropriate verification, review, and approval of design changes before their implementation.
每个制造商都应建立并维护程序,用于在实施设计变更之前进行识别、记录、确认或在适当的情况下验证、审查和批准设计变更。
注意:
以上概述了产品投入生产后发生的设计更改的要求,但是,在设计和开发上市前阶段控制变更也是必要的。 这些更改需要在产品主文档 (DMR) 中进行控制、验证、审查和记录。
本文重点介绍被视为次要性质的更改,以及重大更改将通过 ISO 13485:2016 7.3 中涵盖的设计和开发项目进行的更改
现在,让我们看一下设计变更控制的每个阶段,如上图所示
变更请求:
有许多类型的事件可以触发设计变更,其中一些最常见的事件是:
* 客户的投诉/抱怨,可能引发CAPA或设计变更。
* 流程改进或制造流程变更
* 更新法规或质量管理体系变更
* 标签细微的更改
所有微小的更改都应从记录在案的设计变更请求开始,即工程变更请求 (ECR)。
变更申请表应包括以下内容:
* 请求变更的人员姓名和日期。
* 从质量文件控制团队处获得的变更编号。
* 变更说明
* 变更的原因和理由
* 风险评估,包括对监管要求的任何影响
变更审查和批准:
提交变更请求后,需要通过研发、工程和质量对其进行审查并正式批准,同时记录在文控员的变更请求表上,然后才能正式开始变更过程。
PS:为帮助更好地按理其变更控制,可以考虑选择软件平台来处理变更流程,但具体的平台在此暂时不作推荐。
风险和监管评估:
除了最简单的变更外,应对所有变更进行风险评估,并在设计变更过程开始之前完成。正式的风险评估可以帮助指导设计及其验证和确认计划。对设计变更的审查以及风险评估应包括变更对产品的影响的评估,包括但不限于正在进行开发的产品和已经定型的产品。
根据所请求变更的性质,可能还需要进行监管评估。PS:不同的市场,监管要求可能会有所不同,需要加以考虑。
或者,如果设计变更相当重大,则可能需要通知当前的市场监管机构,甚至在某些情况下,需要先从监管机构获得适当的书面许可后,才能实施变更。注意,这是一个关键因素,因为它可能会对设计变更的实施产生重大影响。
风险和监管评估需要进行记录,包含在变更控制的文档中。
设计变更和验证:
在批准变更请求通过并完成风险评估后,可以继续推进设计变更活动。
在整个变更活动的过程中,可以根据需要组织适当的设计评审。
设计变更的验证和确认根据分析结果执行,并在变更控制文件中记录或引用。
文件更改和批准:
在整个设计变更过程中,做出的有关变更的决策都需要被记录在案。包括更改的原因,包括理由、风险评估和采取的任何行动、更改的详细信息以及执行的验证和确认。
一旦设计变更(包括验证和确认活动,如适用)完成,就可以更新、批准和发布适当的文件,包括产品规格变化、程序修订、BOM修订等(举例),同时准备关闭已完成的工程变更请求(ECR)。
之后,根据最新的质量管理体系文件运行质量控制。

来源:医械研发


