您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-12-12 18:26
一、基本概况
1、自然环境
巴西国土面积851.04 万平方公里,位于南美洲东部,北邻法属圭亚那、苏里南、圭亚那、委内瑞拉和哥伦比亚,西邻秘鲁、玻利维亚,南接巴拉圭、阿根廷和乌拉圭,东濒大西洋。海岸线长约7400公里。国土的80%位于热带地区。
2、人口和行政区划
巴西总人口为 2.03亿,排名拉美第一、世界第七。官方语言为葡萄牙语。全国共分26个州和1个联邦区。州下设市,全国共有5570个市。首都巴西利亚(Brasília),人口385.8万(2022年),是巴西的政治中心。圣保罗是巴西最大的城市,是全国工商、金融、交通中心。里约热内卢是巴西第二大城市,是世界著名的旅游胜地。
3、2024年出口概况
2024年1-6月,中国向巴西出口医疗器械总计约36.93亿人民币,同比增长约9.48% 。
二、巴西医疗器械监管机构和法规要求
巴西国家卫生监督管理局(Agência Nacional de Vigilância Sanitária,ANVISA)负责巴西的医疗器械产品注册,隶属巴西卫生部。ANVISA主要负责所有医疗器械、体外诊断产品及其他健康相关产品(如药品、卫生用品、化妆品等)的上市前与上市后的管控。巴西现行医疗器械法规主要有RDC nº 751 de 15/09/2022(医疗器械)以及RDC nº 830 de 06/12/2023(体外诊断医疗器械),和欧洲的规定较为相似。
三、医疗器械定义
根据法规RDC nº 751, de 15 de setembro de 2022,将医疗器械定义如下:
医疗器械是指由制造商确定将其单独或组合用于人体的,以达到以下任何特定医疗目的的任何仪器、器械、设备、植入物、体外诊断医疗器械、软件、材料或其他物品,其主要预期用途不是通过药理学、免疫学或新陈代谢手段在人体内实现的,而是通过这些方式辅助实现其预期用途:
a) 疾病的诊断、预防、监测、治疗(或缓解);
b) 损伤或残疾的诊断、监测、治疗或修复;
c) 解剖学,生理学或病理学过程或状态的研究、更换、改变;
d) 支持或维持生命;
e) 控制或帮助受孕;
f) 通过对人体样本(包括器官和组织捐献)进行体外检查提供信息。
更多有关有源医疗器械、一次性医疗器械、植入式医疗器械、侵入性医疗器械、体外诊断试剂等定义的详细信息请见:Conceitos e definições
四、产品分类
法规RDC nº 751, de 15 de setembro de 2022附录一,规定了巴西医疗器械的分类规则。根据对人体造成可能风险的高低,将医疗器械被分为四类:Class I-低风险、Class II-中低风险、Class III-中高风险、Class IV-高风险。
温馨提醒:制造商可以与已注册或备案的产品结合查询以确定产品分类,查询清单、数据库。
五、认证流程
1、认证模式
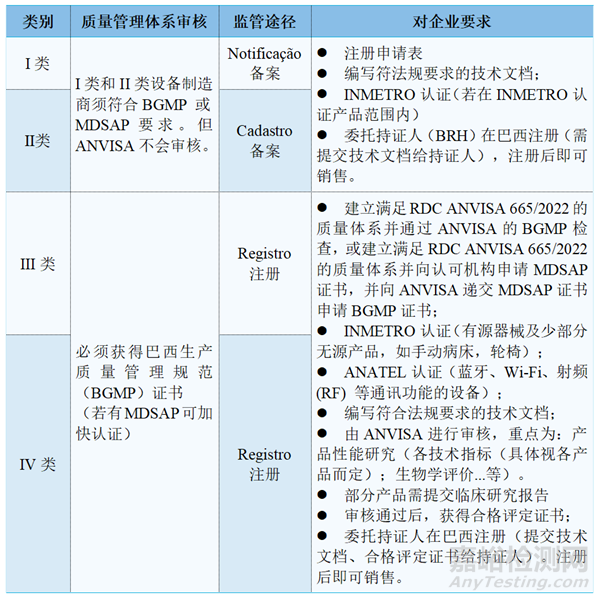
*特别提醒:
1、有关巴西委托持证人BRH的要求
所有境外制造商必须任命一个当地代表作为BRH,也就是我们常说的巴代。因为ANVISA要求只有巴西本地的企业才能提交产品注册申请,并且证书由BRH持有。
2、INMETRO认证
根据RDC 27/2011法规,电子电器类医疗产品必须先取得INMETRO证书。根据现行医疗器械INMETRO认证法规ORD 384, 证书没有有效期的规定,但企业需要接受年度现场监督审核,审核通过后方可维持证书有效性。
3、关于 ANATEL
ANATEL是巴西负责监管该国电信部门的机构。根据第242号决议,具有蓝牙、Wi-Fi、射频 (RF) 等特定功能的设备需要ANATEL认证,即所有发往巴西市场电信链的产品都必须有ANATEL认证。
2、巴西认证流程图示
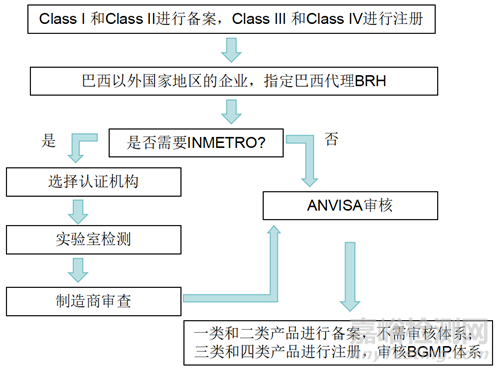
3、注册提交技术文档资料要求
◆ 巴西授权代表协议
◆ 产品说明书及标签
◆ 产品技术规格及图示
◆ 可用性评估报告
◆ 生物相容性评估
◆ 测试报告及一致性声明
◆ 风险评估报告
◆ 软件开发文档
◆ ISO 13485证书或质量手册
◆ 工厂平面图示等
4、注册周期及费用、证书维护及费用
1、注册周期及证书有效期

注:以上预估周期基于非临床试验和临床试验已完成的情况。
2、注册费用
卫生监督检查费(FRVS)的收费不是固定的,根据申请认证的公司的规模以及认证的产品来定,不同公司规模会有不同程度的费用减免。
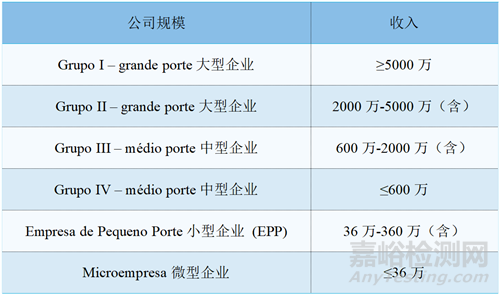
注:收入单位:BRL雷亚尔,参考汇率BRL:CNY=1.00 : 1.26
有关费用的更多详情请见:Saiba o que é e como é cobrada a taxa de fiscalização
3、注册审核费用
◆ Ⅰ类和Ⅱ类产品,ANVISA官方注册费用为240USD。
◆ Ⅲ类产品,ANVISA官方注册费用为970USD。
◆ Ⅳ类产品,ANVISA官方注册费用为1060USD,具体以官方报价为准。
◆ 注册服务费用以巴西代理报价为准。
4、证书维护费用
年度费用以巴西代理报价为准。
5、简化申请流程
2024年4月8日,Anvisa发布了规范指令(IN) 290/2024,为医疗器械注册申请的分析和决定建立了优化程序。
值得注意的是该指令适用于经认可的海外监管机构授权上市的Class III类和Class IV类的医疗器械。有了这一措施,Anvisa在评估已获同等外国监管当局批准的产品时将更加灵活,巩固了在采用监管信任机制方面迈出的一大步。
根据文件,从今年6月3日起,在四个同等外国监管机构(澳大利亚、加拿大、美国和日本)监管的市场授权的医疗器械,可以根据申请公司的声明进行简化分析。
CAPITULO II
DAS AREE RECONHECIDAS
Art. 69 Para fins de adocäo do procedimento otimizado de análise s¤oconsideradas as seguintes AREEe respectivas comprovacöes de registro ou autorizacao:1-Austrália: Australia Therapeutic Goods Administration (TGA)-AustralianRegister of Therapeutic Goods (ARTG);
ll - Canadá: Health Canada (HC)-Medical Device Licence,
1ll-Estados Unidos da América (EUA): US Food and Drug Administration
(US FDA)-510(k) clearance, Premarket Approval (PMA) ou 513(f)(2)"De Novo"; eIV - Jap¤o: Japan Ministry of Health, Labour and Welfare (MHLW)- Pre market approval (Shonin).
为此,必须提交文件,证明运往巴西市场的产品具有经认可的监管当局批准的相同生产特性、适应症和预期用途。允许制造商在申请巴西市场准入时利用澳洲,加拿大,美国,日本四国监管机构的注册,以简化和加快巴西产品注册的过程。
六、有关UDI的要求
2021年12月29日,ANVISA 发布了RDC 591/2021号决议,该决议规定通过医疗器械唯一识别系统 (UDI) 识别向ANVISA通报或注册的医疗器械(医疗产品和体外诊断产品)。不适用于定制医疗器械和临床研究中的医疗器械。RDC 591/2021 描述了医疗器械唯一识别系统 (UDI) 的工作方式,以及分配UDI的截止日期。新决议于2022年1月10日生效。

* 2022年1月,随着这项法规的实施,UDI标签在冠状动脉支架、药物洗脱冠状动脉支架以及髋关节和膝关节置换术中的植入物上成为强制性的。有关详细信息,请参见ANVISA UDI指南。
七、巴西体系要求
1、生产III类和IV类医疗器械或IVD器械的公司必须获得巴西生产质量管理规范(B-GMP)认证证书,以符合巴西GMP质量管理体系要求。巴西RDC第665/2022决议和RDC第497/2021号决议规定了针对医疗器械的要求,这些要求与美国食品药品监督管理局(FDA)的质量体系法规(21 CFR 820部分)和ISO 13485的要求相似。
相关链接:
◆RDC 665/2022 - Regulamento Técnico de Boas Práticas de Fabricação de produtos médicos e produtos para diagnóstico de uso in vitro.医疗产品和体外诊断产品良好生产规范的技术法规。
◆RDC 687/2022 - Critérios para a concessão ou renovação da Certificação de Boas Práticas de Fabricação de Dispositivos Médicos.医疗器械良好生产规范认证授予或更新的标准。
◆RDC 497/2021 - Procedimentos administrativos para concessão da Certificação de Boas Práticas de Fabricação e da Certificação de Boas Práticas de Distribuição e/ou Armazenagem.授予良好生产规范认证和/或良好分销和/或储存规范认证的行政程序。
◆RDC 850/2024 - Altera a Resolução da Diretoria Colegiada - RDC nº 497 de 2021 para ampliar a validade da Certificação de Boas Práticas de Fabricação de fabricantes de dispositivos médicos concedidas por meio do Programa MDSAP para quatro anos.修订合议委员会决议 - 2021 年第 497 号 RDC,将通过 MDSAP 计划授予的医疗器械制造商良好生产规范认证的有效期延长至四年。
2、医疗器械单一审核程序(Medical Device Single Audit Program (MDSAP) )项目是由International Medical Device Regulators Forum (IMDRF,国际医疗器械监管机构论坛)提出,美国(U.S. FDA)、澳大利亚(TGA)、巴西(ANVISA)、加拿大(HC)、日本(MHLW和PMDA)五国的监管机构认可并加入的一套新的审核程序。
◆ MDSAP审核要求指南请见:MDSAP AU P0002 Audit Approach
◆ 有关MDSAP的更多详细信息请见:
1)Medical Device Single Audit Program (MDSAP)
2)Programa de Auditoria Única em Produtos para Saúde – MDSAP (Medical Device Single Audit Program)
其中BGMP, Brazilian Good Manufacturing Practice (BGMP) , RDC 665/2022,与ISO 13485的要求类似。
审核特点:
1)对管理者代表更高的要求,包括具体授权,能力和培训的要求。
2)要求依据各国法规要求,建立产品注册控制流程,代理人协议。
3)核对注册信息(当产品已经在这些国家有销售时)。
4)核对各国法规相关程序文件,比如产品分类、标签制作、符合性申明起草等。
5)核对产品或体系变更通告情况;以及对应程序文件(什么情况下启动通告,如何通告,通告报告等)。
6)对“测量、分析和改进”流程的重视,CAPA。质量数据的获得(包括反馈等生产后信息),不合格调查,分析、纠正和预防的实施。依据巴西ANVISA要求,公司应确定所有咨询方有相关能力和资格。
7)上市后信息收集(顾客投诉等),风险管理。
8)各国不良事件通告。
9)对设计开发的重视,设计开发验证,风险管理。
10)对生产工序审核(Priority criteria for selection),比ISO13485更细的要求,对工艺验证的重视。
11)更为严格的采购控制。

来源:广东医疗器械学会


