您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-07-11 20:14
我们都知道药品的三大基本要素是安全,有效,质量可控。一般情况,安全性是一个药品能上市的必要前提,而杂质则是影响药物安全的重要因素之一,如青霉素过敏等不良反应就是由于杂质引起。
随便翻开哪国药典,检查项除性状、鉴别、溶出、含量外,剩下有关物质、异构体、残留溶剂、重金属等项目都是来检查药物的杂质的,由此可见杂质研究的重要性。
本文通过ICH指导原则中的杂质三限(报告限,鉴定限,界定限)进行简单的解说,浅显讲解新药和仿制药研究中对杂质研究的策略,文中如有误读或理解不准确之处,请及时指出,共同提高。
一、杂质的定义及分类
简而言之,药物的杂质是指任何影响药品纯度的物质。具体包括无机杂质,残留溶剂和有机杂质。
无机杂质:主要来源于生产过程,可能由于试剂,催化剂,配位体等,结构一般已知确定,控制起来较为容易。
残留溶剂:是生产过程中使用的有机或无机溶剂,毒性基本都有记载,因此控制的限度已知,且储存过程中不会增加,研究思路也很明确,控制到规定日剂量限度下是唯一的解决思路。
有机杂质:相比无机杂质和残留溶剂,有机杂质在来源上更为复杂,包括降解产物、反应物残留、副产物、中间体等,结构有已知的也有未知的,生产和储存过程中还有可能会增加,并且说不准哪一个未知杂质就有剧毒。因此有机杂质处理起来也最为棘手,需要用辩证思维来具体情况具体分析。
二、杂质三限及控制策略
凡事皆有度,量变足够才能引起质变。即便是剧毒的杂质也只有到达一定数量,人体耐受不了才会造成危害。因此ICH根据不同的杂质摄入量对人体造成危害的风险级别对杂质设定了3个不同限度,按“低—中-高”风险递增分为:报告限、鉴定限和界定限。
1、报告限
定义:报告限是指需要记录的最低限度。低于这个限度的杂质实在太小了,临床上完全不会有危害,没必要记录,高于这限度的含量也没什么危害,记录下放在那放着即可,没必要知道其结构和做进一步研究。
ICH规定:ICH分别对原料药和制剂中的杂质报告限度做如下规定:
|
—— |
每日主成分最大剂量 |
报告限(%) |
|
原料药 |
≤2g/天 |
0.05% |
|
>2g/天 |
0.03% |
|
|
制剂 |
>1g |
0.05% |
|
≤1g |
0.10% |
表1 ICH对杂质报告限的要求
应对方案:
懒着理你。不管新药还是仿制药,有效期内低于报告限的杂质的存在和我没关系……
小结:
低于报告限的的杂质,不用积分,无视他的存在。高于报告限而低于鉴定限的杂质,不管储存过程中长不长,新药中忽视他,忽视它,仿制药中低于定量限即可,不用管比原研大了还是小了。
2、鉴定限
定义:鉴定限是指需要做杂质结构确证的最低限度。低于此限度的杂质,临床上也不会有什么危害,记录起来放着就可。高于此限度的杂质量已经达到可能对人体造成危害的限度,应对措施自然水涨船高,需做结构确证及后续工作。
ICH规定:ICH分别对原料药和制剂中杂质鉴定限度做如下规定
|
—— |
每日主成分最大剂量 |
鉴定限(取低者) |
|
原料药 |
≤2g/天 |
0.10%或1.0mg/天 |
|
>2g/天 |
0.05% |
|
|
制剂 |
>2g |
0.1%或2mg/d(取低者) |
|
100mg~2g |
0.2%或2mg/d(取低者) |
|
|
10~100mg |
0.2%或2mg/d(取低者) |
|
|
1~100mg |
0.5%或20ug/d(取低者) |
|
|
<1mg |
1.0%或5ug/d(取低者) |
表2 ICH对杂质鉴定限的要求
应对方案:
当产生的杂质超过鉴定限时(低于界定限),根据杂质界定与鉴定决策树(下图)来进行研究,有三种方案选择:
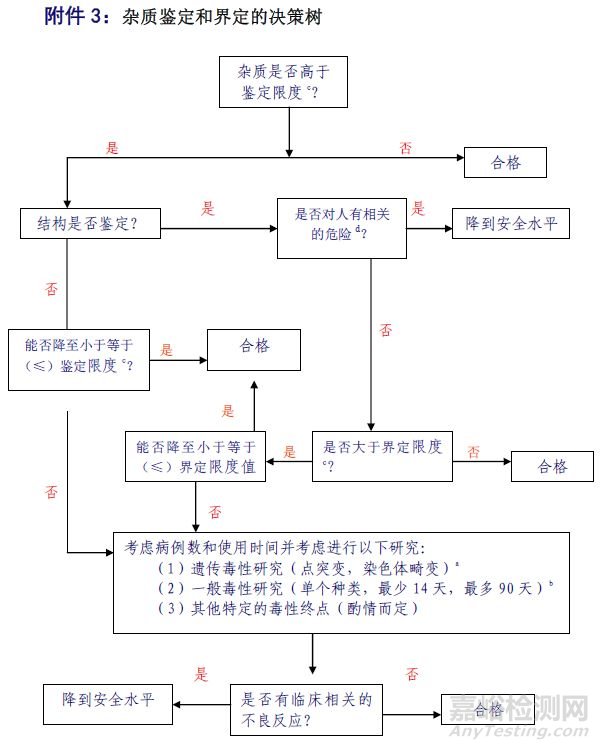
图1:杂质鉴定与界定决策树
方案1:弄清杂质来源,改进工艺将杂质控制在鉴定限以下,避免做杂质鉴定及相关研究。从上表可看出,由于制剂的鉴定限很多情况下比原料宽泛很多,原料的杂质往往没必要做到鉴定限以下。
方案2:弄清杂质结构,通过文献记载来确定是否有毒性,分三种情况来处理:
如果有毒性,需降到毒性杂质限度以下;
如果确认无毒,认为合格,必要情况下甚至可适当放宽限度;
文献没有记载,按合格处理。
方案3:既不做杂质结构确认,也不把杂质降低到鉴定限度以下。那么有一大波试验正向你袭来……这样可能涉及到的试验就多了,研究成本可能也大大增加(这条是针对新药研发,ICH中阐述:对于一个通过充分的安全性研究和临床研究的新药制剂,其中任何一个降解产物的水平被认为是通过界定的——不过这个方案对仿制药来说是通常是找死)。
小结:
ICH尽管对高于定量限杂质给予了三种不同的解决方案和路径,但在做仿制药时第一条路是最佳方案,第二条路备选,第三条则是死路一条。而新药首选第三条方案。
3、界定限
定义:界定限是超越界定限的杂质含量已经达到足以对人造成危害的数量。高于该限度的杂质应进行动物毒理研究,确定临床使用的安全限,且药品中的杂质含量应在效期内不大于该限度。
ICH规定:ICH分别对原料药和制剂中杂质界定限度做如下规定:
|
—— |
每日主成分最大剂量 |
界定限(取低者) |
|
原料药 |
≤2g/天 |
0.15%或1.0mg/天 (取低者) |
|
>2g/天 |
0.05% |
|
|
制剂 |
>2g |
0.15% |
|
100mg~2g |
0.2%或3mg/d(取低者) |
|
|
10~100mg |
0.5%或200ug/d(取低者) |
|
|
1~100mg |
1.0%或50ug/d(取低者) |
|
|
<1mg |
1.0%或50ug/d(取低者) |
表3 ICH对原料药中杂质界定限的要求
应对方案:
当产生的杂质超过界定限时,与鉴定限类似,仍需要根据“降解产物界定与鉴定决策树”来进行研究,但有有所不同。有三种方案选择:
方案1:改变工艺做到界定限以下按照鉴定限处理方式处理,或者直接做到鉴定限以下,尽可能降低杂质量。
方案2:弄清杂质结构,通过文献记载来确定是否有毒性,分三种情况来处理:
如果有毒性,则需降到毒性杂质限度以下;
如果确认无害,则认为合格,必要情况下甚至可适当放宽;
没有记载,要么改进工艺将杂质做到界定限以下,按上述鉴定限杂质处理,要么准备做一大波试验(和鉴定限处理主要的不同之处)。
方案3:和鉴定限处理的第3方案一样,新药逻辑:既不做杂质结构确认也不降低杂质限度,通过安全性研究和临床研究来检验。事实上这种情况即便是没做杂质结构确认,研究者心理也有数的。
小结:
对超过界定限的杂质且原研产品中没有的杂质,对仿制药来说方案2门虽然开着但也仅留个门缝,方案3已经被堵死,唯一的方案是选择方案1。而对新药来说,方案2和方案3对新药仍是很好的选择。
三、结论
通过以上阐述,我们了解到杂质三限的概念及高于三限的处理应对方法:
报告限不用管它,随它去。
鉴定限和界定限处理情况相似并分多种情况。看起来似乎鉴定限处理相对简单,但在实际操作上,鉴定限和界定限处理经常会十分接近甚至重叠,因此对仿制药来说,最优的选择是直接将杂质做到鉴定限以下,而另外两种方案对短平快为特点的仿制药来说,无论是投入的资金成本、时间成本,往往都是仿制药企业无法承受的。
但我们也不能只看到狼吃肉,却不见狼挨揍,相比新药在研发中动辄投入十几亿美元或几十亿美元,以及将近10年的时间来说,多做的那点实验实在不算什么,而且人家研究过的东西可以直接拿来用,很赚了。因此如果真遇到极好的项目,多花点钱做临床和毒理仍然是值得的。

来源:HPC药闻药事


