您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2023-08-02 10:16
本文适用于与静脉输液器具配合使用的一次性使用静脉营养袋。
一、静脉营养袋的基本结构
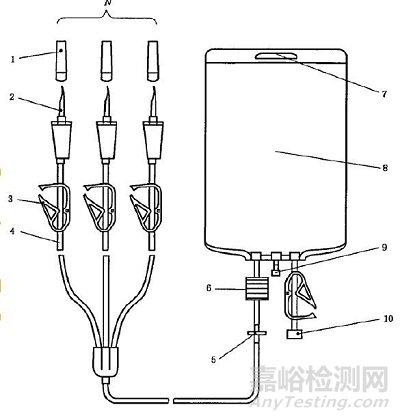



二、一次性使用静脉营养袋的主要风险
按照GB/T 42062《医疗器械风险管理对医疗器械的应用》标准的要求,已识别的风险因素应包括但不局限于以下方面:
1.1原材料的生物学和化学危害
热原
材料或材料来源变化
原材料纯度和可沥滤物
原材料的生物相容性
1.2生产加工过程可能产生的危害
添加剂、助剂、辅剂的残留
生产环境洁净度
微粒污染
内毒素
1.3产品使用风险因素
泄漏
连接件脱落或断裂
染菌
1.4灭菌过程可能产生的危害
灭菌方式对产品不适宜,灭菌不完全、灭菌过程产生的有害物质(如环氧乙烷)等。
1.5不正确使用产生的危害
未按照说明书中操作方法操作,使用过程中产品被微生物、微粒侵入等;重复使用造成患者感染;输注与产品不相容的营养液或药液。
1.6产品包装可能产生的危害
包装破损、标识不清等
三、一次性使用静脉营养袋的性能研究实验要求
1.产品技术要求
产品技术要求应包括但不局限于以下内容:
1.1物理性能
应包含YY/T 0611《一次性使用静脉营养输液袋》适用的相关性能,产品的结构非YY/T 0611的设计结构,或在YY/T 0611给出的结构基础上还有其它设计元素的,应制订与之相关的性能要求,如进液管路上装配的药液过滤器等。
1.2化学性能
应包含YY/T 0611适用的化学要求。
1.3 其他
无菌、细菌内毒素。
产品性能指标和试验方法若不适用于YY/T 0611的相关要求。开发人自行制定性能要求的试验方法的,应在研究中补充方法学验证研究。
2.原材料控制
常见的静脉营养袋的贮液袋的原材料为以乙烯单体和乙酸乙烯为主要原料聚合而成的乙烯-乙酸乙烯酯共聚物(EVA),贮液袋为EVA材质的,开发人应明确EVA的成分及占比信息、残留单体的控制信息以及原材料的质控标准。
采用其他高分子材料制成的静脉营养袋,应明确原材料的选择依据及来源,建议选用已有相关人类临床应用史的原材料,说明原材料与已上市同类产品原材料的异同及性能对比情况。
明确产品部件所用材料的化学名称、商品名/牌号、化学结构式/分子式、供应商名称、符合的标准等基本信息。应明确所用原材料的质控标准,对原材料进行检验和相关验证研究,确保符合相应标准。
对于首次应用于医疗器械的新材料,应开展该材料适合用于人体预期使用部位的相关研究。
产品初包装采用的材料应能保证产品在灭菌、贮存和运输过程中对产品性能和安全性不产生不利影响。应明确初包装材料的来源、质量控制标准并进行验证研究。
3.产品性能研究
应当开展产品性能研究,明确包括有效性、安全性指标的确定依据、所采用的标准或方法、采用的原因。
3.1物理性能研究至少包括:
产品的微粒污染指数、瓶塞穿刺器的性能、可拆开式管路连接件的性能、泄漏性能、各连接件之间的连接牢固度、止液夹的止液性能、进气器件(若有)的性能、贮液袋的性能、注射件(若有)的性能、防重开启截留装置的性能(若有)、输液器插口的性能等。
3.2化学性能研究:
对与营养液直接接触部分的聚合物建议根据材料特性,开展产品的化学性能研究,至少包括浸提液还原物质、金属离子、酸碱度、蒸发残渣、紫外吸光度、环氧乙烷残留量(如适用)等。
4.生物相容性研究
按GB/T 16886系列标准规定的要求进行评价,本产品为外部接入器械,与人体接触部位为血路间接接触,接触时间为不大于24h的短期接触,应评价的项目包括:热原、细胞毒性、致敏反应、皮内反应、急性全身毒性、血液相容性。
5.灭菌工艺研究
5.1应明确灭菌工艺(方法和参数)及其选择依据,并开展产品灭菌方法适宜性的验证研究,同时开展选用的灭菌方法可以使产品达到的无菌保证水平(SAL)的灭菌确认研究,产品的无菌保证水平(SAL)应不低于10-6。
5.2残留毒性:若产品灭菌采用的方法容易出现残留,如环氧乙烷灭菌,应开展研究明确残留物信息及采取的处理方法。
6.产品货架有效期和包装研究
6.1货架有效期
产品货架有效期的研究可参照《无源植入性医疗器械稳定性研究指导原则》,按照产品实际情况执行。医疗器械货架有效期包括产品有效期和包装有效期,货架有效期的验证试验类型通常可分为加速稳定性试验和实时稳定性试验两类。
加速稳定性试验的具体要求可参考YY/T 0681系列标准,在进行加速稳定性试验研究时应注意:产品选择的环境条件的老化机制应与宣称的运输储存条件下真实发生的产品老化的机制相匹配。对于在加速稳定性试验研究中可能导致产品变性而不适于选择加速老化试验方法的,应以实时稳定性试验进行测定和验证。实时稳定性试验中,开发人应根据产品的实际生产、运输和储存情况确定适当的温度、湿度、光照等条件,在设定的时间间隔内对产品进行检测。
6.2包装及包装完整性
在宣称的有效期内以及运输储存条件下,保持包装完整性的依据。开发人应开展产品有效期内的包装验证和运输验证。产品包装验证可依据有关国内外标准(如GB/T 19633系列标准等)进行。
7.产品的药物相容性评价
静脉营养袋与药物的相容性试验应考察其与药物之间是否会引起相互的或单方面的迁移、吸附以及质量的变化,包括物理相容性、化学相容性等多方面内容。本试验应在较恶劣的或模拟临床使用条件下进行,以研究药物与静脉营养袋之间的影响。
7.1药物相容性试验应考虑以下方面:
7.1.1生产所用材料;
7.1.2添加剂、加工过程的残留物、单体、起始物质;
7.1.3降解产物;
7.1.4药物与营养袋的相互作用;
7.1.5试验用药物的物理和化学性质。
7.2药物相容性试验的要求:
7.2.1药物试验
本试验考察药物通过营养袋前后质量的变化情况和营养袋对药物的吸附作用,建议选择预期拟输注的药物分别进行研究。
7.2.1.1试验用药物溶液的浓度应采用与临床使用浓度一致或使用更高浓度的药物溶液,并保证药物溶液与营养袋有足够的接触时间。按照试验药物的质量标准检测通过营养袋前后药物溶液的理化指标,综合考察药物通过营养袋前后的质量变化。
7.2.1.2 药物吸附试验应考察营养袋内相同体积的药物溶液,在不同时间周期药物溶液被营养袋吸附的情况。
7.2.2添加剂、残留单体、降解产物等的溶出和迁移
建议选用可代表临床营养成分配方的营养液开展迁移研究。通过模拟临床实际使用状况,考察在规定温度条件下,经过一定的时间周期接触后,通过光谱法、色谱法等可行的测定方法测定样品中添加剂、残留单体、降解产物等的溶出和迁移情况,并对溶出物质进行毒理学评估。测定方法需进行方法学验证。
7.2.3温度
由于物质在高温状态下的迁移速度要高于常温或低温状态,药物试验和迁移试验可考虑在40℃±1℃温度条件下,考察药物溶液与营养袋接触后的相互变化及营养袋添加剂的迁移情况;若用于验证的药物溶液不耐高温,可考虑在常温(25℃±1℃)下试验。研究需采用经过方法学验证的方法进行。
8.其他
静脉营养袋已列入《免于进行临床评价医疗器械目录》(以下简称《目录》),对于符合《目录》中分类编码为14-02-11的,开发人需将产品相关信息与《目录》所述内容进行对比,具体要求可参照《列入免于进行临床评价医疗器械目录产品对比说明技术指导原则》。

来源:嘉峪检测网


